何首乌药效成分3种提取方法比较
2016-12-21杨劝生
杨劝生
甘肃天水岐黄药业有限责任公司,甘肃 天水 741018
何首乌药效成分3种提取方法比较
杨劝生
甘肃天水岐黄药业有限责任公司,甘肃 天水 741018
目的:研究中药材何首乌的不同提取方法,比较所提取的成分,探索优化何首乌的提取工艺。方法:采用高效液相色谱法(H PLC)分析测定何首乌渗漉提取、75℃回流提取、89℃回流提取的提取液的二苯乙烯苷、蒽醌类化合物含量,并进行3种不同提取方法的提取液化学成分指纹图谱相似度及所得干物质考察。结果:3种提取方法提取的二苯乙烯苷、蒽醌类化合物的含量基本接近。结合蒽醌的含量89℃回流提取略高于渗漉提取;75℃回流提取略低于渗漉提取。何首乌3种不同提取方法制备的供试品溶液的特征指纹图谱与共有模式的相似度均大于0.95,符合国家对药材指纹图谱研究的技术要求(大于0.9)。结论:3种不同提取方法制备的指纹图谱相似程度高,其提取液化学成分差异不大。
何首乌;高效液相色谱法;渗漉;回流;指纹图谱
苁蓉通便口服液是甘肃天水岐黄药业有限责任公司独家研制的专利产品,以肉苁蓉、何首乌、枳实、蜂蜜科学组方,具有滋阴补肾,润肠通便的功效。其处方中何首乌可解毒消痈,润肠通便,主要成分有二苯乙烯苷类、蒽醌类、磷脂类化合物,以二苯乙烯苷类成分含量最高,其次为蒽醌类成分[1]。本文在何首乌提取采用溶媒及用量一致的情况下,比较回流法和渗漉法提取液的化学成分相似程度、含量等,为苁蓉通便口服液生产中何首乌回流提取替代渗漉提取提供依据。
1 材料与方法
1.1 仪器与试药 LC-2010A高效液相色谱仪(日本岛津公司提供)。对照品:2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品(批号:110844-200908)、大黄素(批号:110756-200110)、大黄素甲醚(批号:110758-201013)等由中国药品生物制品检定所提供。乙腈为色谱纯,试剂与试药为分析纯,水为重蒸馏水。
1.2 色谱条件[1-2]色谱柱:Dikma C18(4.6 mm× 250 mm,5μm)。经过对下列流动相的选择、对比实验:甲醇-0.5%醋酸溶液(42∶58)、乙腈-水(15∶85)、甲醇-0.1%磷酸溶液梯度、乙腈-0.1%甲酸溶液,以乙腈-0.1%甲酸溶液为流动相梯度洗脱分离效果最佳,峰形及保留时间均比较理想;流速:1.0 mL/min;检测波长为280 nm;柱温30℃;时间72分钟,见表1。
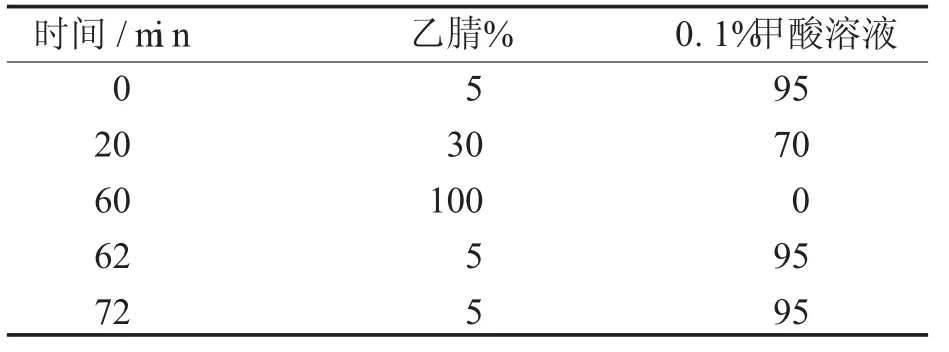
表1 流动相梯度洗脱条件
2 方法与结果
2.1 何首乌药材3种不同提取方法提取液的制备 取何首乌粗粉60g,精密称定,装入内径2.5cm的渗漉管中,加95%的乙醇适量浸渍12小时,室温用9倍量70%乙醇作溶剂,缓缓渗漉,收集漉液,回收乙醇,浓缩,用70%乙醇定容于500 mL的量瓶。取何首乌粗粉60g,精密称定,用9倍量70%乙醇作溶剂,分3次,75℃回流提取,合并提取液,取上清液,回收乙醇,浓缩,用70%乙醇定容于500 mL的量瓶。取何首乌粗粉60 g,精密称定,用9倍量70%乙醇作溶剂,分5次,89℃回流提取,合并提取液,取上清液,回收乙醇,浓缩,用70%乙醇定容于500 mL的量瓶。
2.2 对照品溶液的制备 精密称取2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品适量,精密加入70%乙醇,制成含2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷537.102 μg/mL的溶液,作为对照品溶液。精密称取大黄素对照品适量,精密加入70%乙醇,制成含大黄素78.8 μg/mL的溶液,作为对照品溶液。精密称取大黄素甲醚对照品适量,精密加入70%乙醇,制成含大黄素甲醚24.1516 μg/mL的溶液,作为对照品溶液。
2.3 供试品溶液的制备
2.3.1 供测二苯乙烯苷、测游离蒽醌用 分别精密吸取3种何首乌药材不同提取方法和条件提取液1 mL,置10 mL量瓶,加入70%乙醇至刻度,摇匀,滤过,取续滤液,作为供试品溶液A。
2.3.2 测总蒽醌用 分别精密吸取何首乌药材3种不同提取方法和条件提取液5 mL,置具塞锥形瓶中,水浴蒸干,精密加8%盐酸溶液20 mL,超声处理(功率100 W,频率40 kHz)5分钟,加三氯甲烷20 mL,水浴中加热回流1小时,取出,立即冷却,置分液漏斗中,用少量三氯甲烷洗涤容器,洗液并入分液漏斗中,分取三氯甲烷液,酸液再用三氯甲烷振摇提取3次,15 mL/次,合并三氯甲烷液,回收溶剂至干,残渣加70%乙醇使溶解,转移至10 mL量瓶中,加70%乙醇至刻度,摇匀。精密吸取上述定容液1 mL,转移至10 mL量瓶中,加70%乙醇至刻度,摇匀滤过,取续滤液,作为供试品溶液B。
2.4 理论板数的计算 在上述条件下,将对照品溶液注入高效液相色谱仪中,记录色谱图,量出供试品溶液2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷峰的保留时间Tr(以分钟或长度计算)和半峰高宽 (Wh/2),按n=5.54(Tr/Wh/2)2计算色谱柱的理论板数。理论板数按盐酸2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷峰计算不低于2 500。
2.5 分离度 依据高效液相色谱法(《中国药典》2010年版一部附录Ⅵ D)的规定,待测峰与其他峰的分离度应不小于1.5,按分离度的计算公式计算供试品色谱中2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷的峰与其他峰的分离度,供试品的分离度均符合规定。
2.6 精密度 分别精密吸取2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品溶液、大黄素对照品溶液各10 μL,按上述色谱条件测定,各进样5次,测定峰面积积分值,RSD分别为0.13%、0.86%、0.12%。
2.7 线性关系考察 精密吸取上述2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品溶液4、6、8、10、12、14 μL,按上述色谱条件分别注入液相色谱仪,以对照品的进样量为纵坐标,相应的峰面积积分值为横坐标,计算回归方程:C=6.703 75× 10-7A-0.433 20,r=0.999 2;2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷在2.148 4~7.519 4 μg内有良好的线性关系。精密吸取上述大黄素对照品溶液1、2、4、6、8、10 μL,按上述色谱条件分别注入液相色谱仪,以对照品的进样量为纵坐标,相应的峰面积积分值为横坐标,计算回归方程:C=2.686 04×10-7A-0.031 51,r=0.999 88;大黄素在0.788~7.880 μg内有良好的线性关系。精密吸取上述大黄素甲醚对照品溶液4、6、8、10、12、14 μL,按上述色谱条件分别注入液相色谱仪,以对照品的进样量为纵坐标,相应的峰面积积分值为横坐标,计算回归方程:C=1.38410×10-6A-0.17390,r=0.999 2;大黄素甲醚在0.096 6~0.338 1 μg内有良好的线性关系。
2.8 耐用性 为了保证测定方法的耐用性,我们对供试品溶液的稳定性进行了研究。取同一供试品溶液,分别在0、2、4、6、8小时,按上述色谱条件各进样一次,10 μL/次,结果见表 2。表明2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷、大黄素、大黄素甲醚在8小时内稳定。RSD分别为0.14%、0.89%、0.99%。
2.9 不同提取方法有效成分含量测定 分别精密吸取上述供试品溶液A、供试品溶液B,按上述色谱条件进样,10 μL/次,测定2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷、大黄素、大黄素甲醚积分值,计算。见表2。
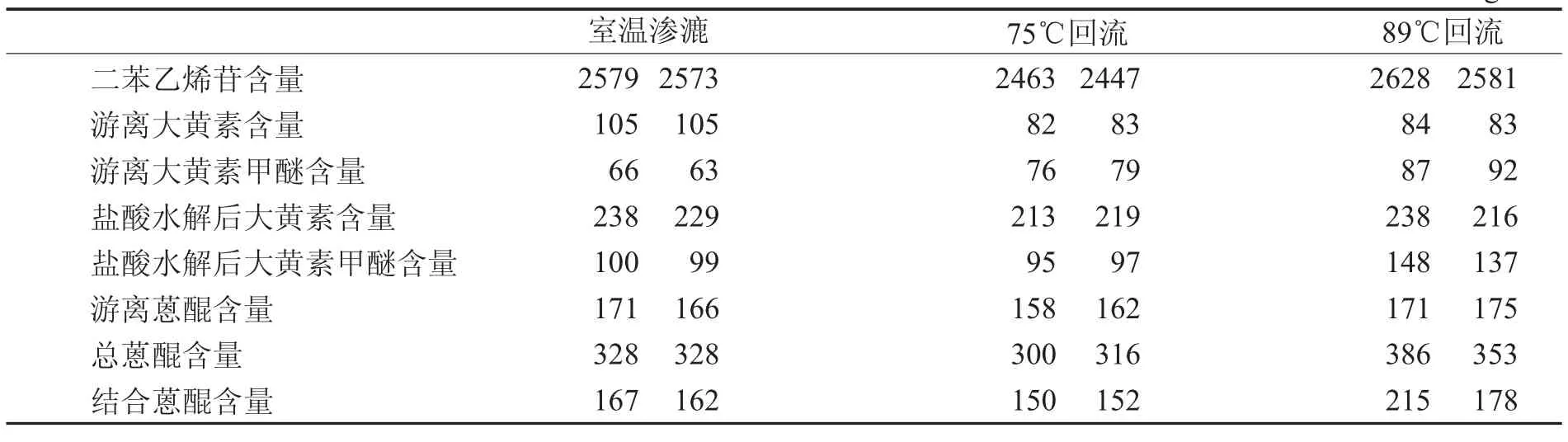
表2 不同提取方法有效成分含量比较 m g
2.10 指纹图谱相似度考察 分别精密吸取何首乌药材3种不同提取方法提取的提取液1 mL,置10 mL量瓶,加入70%乙醇至刻度,摇匀,滤过,取续滤液,作为供试品溶液。
2.1 0.1 方法学考察 按照下列公式计算各色谱峰的相对保留时间及相对峰面积:
相对保留时间=各色谱峰保留时间/二苯乙烯苷色谱峰保留时间
相对峰面积=各色谱峰峰面积/二苯乙烯苷色谱峰峰面积
2.1 0.2 精密度实验 取同一供试品溶液,按上述色谱条件,进样6次,10 μL/次,以二苯乙烯苷色谱峰(6号峰)为参照峰,计算各共有峰的相对保留时间和相对峰面积。相对保留时间RSD为0.06%~0.34%,相对峰面积RSD为0.71%~11.47%。
2.1 0.3 共有模式的建立和相似度计算 将何首乌3种不同提取方法制备的供试品溶液各10份的高效液相特征图谱的结果,导入“中药色谱指纹图谱相似度评价系统A版”,采用“均数法”导出共30份样品的共有模式,见图1。

图1 何首乌三种不同提取方法和条件的供试品溶液各10批的高效液相特征图
3 讨论
何首乌中的主要成分有二苯乙烯苷类、蒽醌类、磷脂类化合物;以二苯乙烯苷类成分含量最高,其次为蒽醌类。现代药理实验表明何首乌中的结合蒽醌类物质,具有促进胃肠蠕动、泻下的作用[3-4]。
对从何首乌3种不同提取方法提取的含量结果和指纹图谱相似度进行分析,发现二苯乙烯苷、蒽醌类化合物的含量基本接近。结合蒽醌的含量以89℃回流提取略高于渗漉提取;75℃回流提取由于提取液的溶媒用量少于渗漉提取,结合蒽醌的含量略低于渗漉提取。何首乌3种不同提取方法制备的供试品溶液的特征指纹图谱与共有模式的相似度均大于0.95,符合国家对药材指纹图谱研究的技术要求(大于0.9)。说明3种不同提取方法制备的指纹图谱相似程度高,由此可以得出3种不同方法提取的何首乌,由于提取溶媒、溶媒用量一致,其提取液的化学成分相似程度高、含量测定结果无明显差异,回流提取没有造成遇热易破坏成分的损失。可认为何首乌回流与渗漉提取比较,其工艺和化学成分没有质的变化,为何首乌回流提取替代渗漉提取提供了依据。
[1] 罗文,刘斌,王伟,等.何首乌药材H PLC指纹图谱研究[J].北京中医药大学学报,2008,31(8):557-560.
[2] 蔡丽芬,张倩,钟国跃.H PLC法同时测定何首乌中二苯乙烯苷和蒽醌类成分的方法学研究[C].北京:全国第8届天然药物资源学术研讨会论文集,2008:337-341.
[3] 罗瑞芝,贾伟.何首乌研究进展[J].中草药,2005,36(7): 1097-1099.
[4] 刘振丽,宋志前.何首乌炮制过程中结合蒽醌含量与泻下作用变化的相关性分析[C].济南:现代化中药制剂发展与中药药理学研究交流会,2007:192-194.
Comparison of Three Extract Methods for the Medicinal Composition of HeShouWu
YANG Quansheng
Qihuang Limited Liability Company of medicine from Tianshui,Gansu Province,Tianshui 741018,China
Objective:To study the different extract methods of herb HeShouWu'compare extracted components,and explore as well as optimize the extraction process of HeShouWu.Methods:The HPLC method was used to analyze and determine the contents of stilbene glycoside,anthraquinones in HeShouWu extract by percolation extraction,reflux extraction at the temperatures of 75℃and 89℃.The similarities and dry matters of the fingerprint of chemical components from extracts by three different extract methods were investigated.Results:The contents of stilbene glycoside and anthraquinones extracted by three extract methods were approximately equal.Contents of combined anthraquinones by 89℃ reflux extraction were slightly superior to percolation extraction;Contents of combined anthraquinones by 75℃reflux extraction were slightly lower than percolation extraction.The characteristic fingerprint and similarity of common patterns of HeShouWu test solution prepared by three different extract methods were all greater than 0.95 which satisfied the technical requirements for the research of the fingerprint of Chinese medicinal herbs(>0.9).Conclusion:The similarity degree of fingerprints prepared by three different extract methods is high and there is not an obvious difference in the chemical components from its extract.
HeShouWu;HPLC;percolation;reflux;fingerprint
R284.2
A
1004-6852(2016)08-0035-03
2015-12-16
杨劝生(1974—),男,副主任中药师。研究方向:药品生产、质量保证与质量控制、药品注册、新药研发。
