84例强直性肌营养不良患者临床与病理分析
2016-12-19蒲传强张羽彤王慧芳毛燕玲刘洁晓
陈 曦, 蒲传强, 张羽彤, 班 瑞, 王慧芳,3, 陈 婷, 石 强, 毛燕玲, 刘洁晓
84例强直性肌营养不良患者临床与病理分析
陈 曦1,2, 蒲传强1, 张羽彤1,2, 班 瑞1,2, 王慧芳1,3, 陈 婷1,2, 石 强1, 毛燕玲1, 刘洁晓1
目的 探讨强直性肌营养不良患者的临床及肌肉病理特点。方法 选择2003~2015年经临床表现、肌电图检查明确诊断为强直性肌营养不良的84例患者为研究对象,行开放式肌肉活检术以获取骨骼肌标本,标本经冰冻切片后进行多种酶组织化学染色并于光镜下观察病理变化特点。结果 在84例患者中,71例患者出现肌强直,57例肌无力,39例肌萎缩,11例白内障。光镜观察发现,5例患者肌肉组织形态学正常,79例患者有形态学异常,其中62例表现为肌纤维大小不等,78例出现肌纤维萎缩,59例可见大量核内移现象,29例可见肌浆块;17例可见肌纤维坏死和吞噬现象。出现肌强直症状与未出现肌强直症状的两组患者间年龄差异具有高度统计学意义(P=0.008)。结论 强直性肌营养不良以肌无力,肌强直,肌萎缩,白内障为其主要临床表现,核内移及肌浆块为其肌肉组织最具特征性的病理改变。
强直性肌营养不良; 肌肉活检; 病理学; 肌浆块
强直性肌营养不良(myotonic dystrophy,DM)是一种累及多系统的常染色体显性遗传病,全球发病率约为1∶20000,任何年龄均可发病,男性多于女性,主要症状为肌无力、肌强直、肌萎缩、早发性白内障、秃发,还可以有心脏传导异常、球麻痹、认知及行为改变等[1]。该病临床表现个体间差异较大,肌肉活检为其诊断的重要方法之一,本文总结分析84例强直性肌营养不良患者的临床与病理特点,以加强对该病认识。
1 材料与方法
1.1 研究对象 2003年9月~2015年9月因疑似为肌肉病而在我科行肌肉活检的2073例患者,经临床表现、肌电图检查和肌肉酶组织化学技术符合强直性肌营养不良诊断的患者84例。
1.2 研究方法
1.2.1 病例资料 收集这些患者的人口学资料、临床症状与体征、个人史与家族史、血清肌酶、甲状腺功能、心电图、心脏超声、肌电图、肌肉酶组织化学结果等资料。
1.2.2 肌活检病理检查 84例患者均于签署知情同意书后行开放式肌肉活检术以获取肌肉标本,标本经液氮预先冷却的异戊烷冻结、冰冻切片(5 μm)后进行酶组织化学染色并于光镜下观察病理变化。染色方法包括苏木精-伊红染色(HE)、改良Gomori染色(MGT)、还原型辅酶I染色(NADH-TR)、琥珀酸脱氢酶染色(SDH)、非特异性酯酶染色(NSE)、三磷酸腺苷酶染色(ATPase)、高碘酸Schiff反应染色(PAS)、油红O染色(ORO)及苏丹黑染色(SBB)。
2 结 果
2.1 一般资料 在2073例肌病患者中,强直性肌营养不良共84例,占同期肌活检总数的4.05%,其中,男性58例(69.05%),女性26例(30.95%)。就诊年龄11~62岁,平均33.37±13.11岁;发病年龄2~54岁,平均24.25±13.61岁,其中男性平均年龄21.86±13.03岁,女性平均年龄29.58±13.63岁,两者间差异具有统计学意义(P=0.015<0.05)。病程0.5~37 y,平均7.0±7.75 y(中位数±四分位数间距)。本组中44例(52.38%)患者为散发,40例(47.62%)患者有明确的家族遗传史。
2.2 首发症状 在84例强直性肌营养不良患者中,44例(52.38%)患者首发症状为肌无力,32例(38.10%)为肌强直,4例(4.76%)为行走缓慢,2例(2.38%)为言语不清,1例(1.19%)为吞咽困难,1例(1.19%)为记忆力减退。
2.3 临床表现 见表1。在84例强直性肌营养不良患者中,共71例(84.52%)患者出现肌强直,主要表现为握拳后松开困难,57例(67.86%)出现肌无力,39例(46.43%)出现肌萎缩,8例(9.52%)出现肌痛,28例(33.33%)患者出现球麻痹;确诊为白内障患者共11例(13.10%),另有2例(2.38%)出现视力下降,2例(2.38%)伴有复视;15例(17.86%)患者出现秃发,其中男性14例,女性1例。5例(5.95%)患者出现心脏受累,其中心律失常4例(4.76%),心力衰竭1例(1.19%);2例(2.47%)患者累及消化系统,表现为便秘1例(1.19%)和腹泻1例(1.19%)。查体发现22例(26.19%)患者叩击性肌球征阳性,26例(30.95%)呈典型“斧状脸”,11例(13.10%)患者出现肌张力改变,其中10例(11.90%)患者肌张力减低,1例(1.19%)患者肌张力增高。18例(21.42%)患者呈对称性腱反射减弱,10例(11.90%)患者腱反射消失。1例(1.19%)患者病理征阳性。
2.4 血清肌酶水平 75例患者检测肌酸激酶(CK)含量,范围41.8~2422 U/L,平均264.80±118.2 U/L(中位数),其中24例(32.00%)正常,33例(44.00%)升高2倍以下,11例(14.67%)升高2~5倍,7例(9.33%)升高5倍以上。75例检测血清乳酸脱氢酶(LDH)含量,范围95.1~521.7 U/L,平均227.12±67.78 U/L。70例检测血清谷丙转氨酶(GPT),范围8.8~132.6 U/L,平均36.55±22.04 U/L。74例检测血清谷草转氨酶(GOT),范围6.2~512 U/L,平均29.70±16.25 U/L(中位数)。
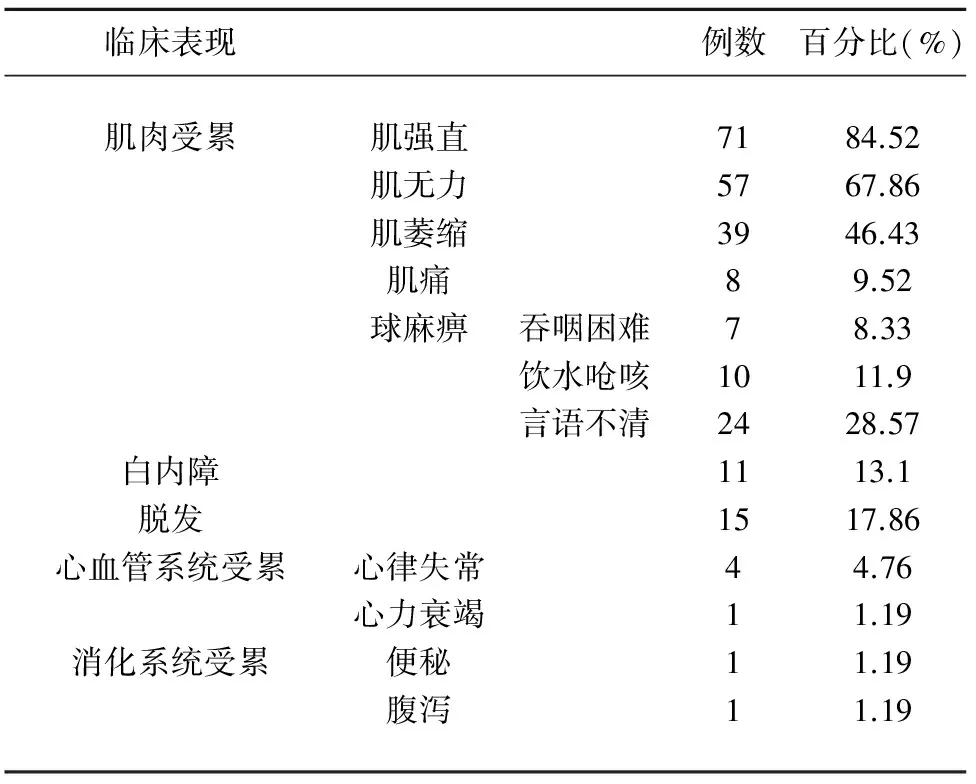
表1 强直性肌营养不良患者临床表现分布
2.5 肌电图结果 在84例强直性肌营养不良患者中共81例接受肌电图检查,其中72例(88.89%)呈现肌源性损害,76例(93.83%)出现肌强直电位,1例(1.23%)出现感觉神经传导速度减慢。
2.6 肌肉活检病理结果 在84例强直性肌营养不良患者中,5例(5.95%)患者肌肉组织形态正常,79例(94.05%)患者肌肉组织形态异常。光镜下观察79例患者肌肉组织病理改变如下:(1)HE染色:62例(78.48%)可见肌纤维大小不等,78例(98.73%)出现肌纤维萎缩,萎缩肌纤维呈小圆形、小角形、不规则形等;51例(64.56%)可见肥大肌纤维,59例(74.68%)可见核内移现象,13例(16.46%)可见核聚集,29例(36.71%)可见肌浆块,21例(26.58%)可见分裂肌纤维,12例(15.19%)可见肌纤维变性,17例(21.52%)可见肌纤维坏死,15例(18.99%)可见结缔组织增生,所有病例均未见炎性细胞浸润。(2)MGT染色:9例(11.39%)可见破碎红纤维,提示出现异常增多的线粒体。2例(2.53%)可见镶边空泡。(3)ATP酶染色:53例(67.09%)患者两型肌纤维均受累,22例(27.85%)仅I型肌纤维受累,4例(5.06%)仅Ⅱ型肌纤维受累;38例(48.10%)患者肌纤维分型良好,8例(10.13%)Ⅰ型肌纤维占优势,33例(41.77%)Ⅱ型肌纤维占优势;11例(13.92%)出现肌源性群组化现象。(4)NADH-TR染色:4例(5.06%)可见轮状纤维,6例(7.59%)可见虫蚀样纤维,2例(2.53%)可见靶纤维。(5)ORO染色:仅1例(1.27%)可见少数肌纤维呈阳性,提示脂质代谢异常。(6)PAS染色:仅3例(3.80%)可见部分肌纤维呈阳性,提示糖原代谢异常。
2.7 有无肌强直症状患者间临床特点比较 在本组84例患者中,共71例(84.52%)患者出现肌强直症状,其余13例(15.48%)患者临床上未出现肌强直症状。统计发现年龄在两组患者间差异具有高度统计学意义(P=0.008<0.01),出现肌强直患者组平均年龄31.77±12.71岁,低于未出现肌强直患者组平均年龄42.08±12.17岁。其余各项包括性别、发病年龄、病程、各项临床症状、血清肌酶水平及肌电图结果在两组间差异均无统计学意义。
3 讨 论
强直性肌营养不良(DM)是一种外显率极高的常染色体显性遗传病,任何年龄均可发病,核心症状为肌无力、肌强直、肌萎缩及早发型白内障[1,2]。肌强直多表现为用力握拳后松开困难和肌球征阳性,可累及面肌、舌肌出现球麻痹症状。秃发、心脏传导异常等均为本病常见症状。中枢神经系统受累者可致认知障碍和行为异常。患者还可出现代谢功能异常,性功能减退,胃肠道症状及周围神经损害等,肿瘤发病风险增高[3]。肌电图检查多可见肌肉强直样放电且呈现短时限、多相波等肌源性损害特点。肌肉活检多可见肌纤维大小不等,大量核内移肌纤维以及肌浆块为其特征性病理改变。
本组84例患者中,以肌无力为首发症状者最为多见,共44例(52.38%),其次为肌强直32例(38.1%)。在本组患者中,共有13例患者临床上未出现肌强直症状,据此将患者分为两组比较临床特点,发现出现肌强直患者组年龄低于未出现肌强直患者组,年龄在两组患者间差异具有高度统计学意义。
肌肉活检为强直性肌营养不良的重要诊断方法。在本组84例患者中,5例患者肌肉组织形态正常,考虑该5例患者病史均在10年以下,尚处在病程早期,且强直性肌营养不良为离子通道疾病,存在光镜下无法识别肌膜异常。其余79例患者肌肉病理改变主要表现为骨骼肌的退行性改变,62例患者出现肌纤维大小不等,78例(98.73%)可见萎缩肌纤维,多呈圆形或小角形,51例患者出现代偿性肥大肌纤维。
核内移为DM患者最为常见且具有特征性的病理改变,本组59例患者可见核内移肌纤维,主要表现为肌核数量增多,高频率出现中心核,纵切面表现为延伸可达数个肌节的链状核,与此前报道类似[4]。
肌浆块为强直性肌营养不良的特征性病理表现,一旦出现即可确诊为DM,依据国内外报道,其检出率仅为20%~50%,且多出现于中晚期病例,本组84例患者中,共有29例患者出现肌浆块,占全部患者的34.52%,与此前报道相似[5~7]。肌浆块在HE染色中呈现紫红色,多位于肌内膜下,在NADH-TR、NSE、ACP染色中浓染提示肌浆块酶活性升高。
13例患者出现核聚集,其中11例患者病程超过10年。4例伴有轮状纤维患者病程均在10年以上,21例可见肌纤维分裂,肌纤维坏死较少见,少数患者出现轻度结缔组织增生,均无炎性细胞浸润。多数病例存在肌纤维分布异常,多为Ⅱ型肌纤维占优势,与国内外报道相似[8,9]。11例呈现肌源性群组化现象,占全部患者的13.10%,明显低于此前文献报道(11/14)[6]。9例患者MGT染色可见破碎红纤维(RRF),提示线粒体的异常增多,考虑为继发性改变,另据汪伟报道,肌纤维的核内移以及破碎红纤维的出现这2种病理改变同肌无力的关系密切[8]。少数晚期病例报道存在肌肉的纤维化、脂质及糖原代谢异常[10],本组84例患者中仅1例提示脂质代谢异常,3例提示糖原代谢异常。
综上所述,肌肉活检对于强直性肌营养不良患者的明确诊断具有重要意义,肌浆块及核内移作为该病特征性病理改变,其临床意义尚有待进一步研究。
[1]Meola G,Cardani R. Myotonic dystrophies:an update on clinical aspects,genetic,pathology,and molecular pathomechanisms[J]. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease,2015,1852(4):594-606.
[2]Udd B,Krahe R. The myotonic dystrophies:molecular,clinical,and therapeutic challenges[J]. The Lancet Neurology,2012,11(10):891-905.
[3]Mueller CM,Hilbert JE,Martens W,et al. Hypothesis:neoplasms in myotonic dystrophy[J]. Cancer Causes & Control,2009,20:10.
[4]Vihola A,Bassez G,Meola G,et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2[J]. Neurology,2003,60(11):1854-1857.
[5]Pongratz D,Schultz D,Koppenwallner C,et al. [Diagnostic value of muscle biopsy findings in myotonic dystrophy (Curschmann-Steinert)(author’s transl)][J]. Klinische Wochenschrift,1979,57(5):215-224.
[6]赵晓萍,蒲传强,吴卫平,等. 强直性肌营养不良症患者肌肉病理学特点研究[J]. 中国现代神经疾病杂志,2006,5(6):389-392.
[7]Meola G. Clinical aspects,molecular pathomechanisms and management of myotonic dystrophies[J]. Acta Myologica,2013,32(3):154.
[8]汪 伟,刘 祺,王朝霞,等. 强直性肌营养不良 I 型患者的临床评分与病理改变的相关性[J]. 中华医学杂志,2013,93(7):508-511.
[9]Nadaj-Pakleza A,Usakowska A,Suek-Pi,et al. Muscle pathology in myotonic dystrophy:light and electron microscopic investigation in eighteen patients[J]. Folia Morphologica,2011,70(2):121-129.
[10]吴筠凡,周志华,韩咏竹,等. 强直性肌营养不良临床、电生理和肌肉病理研究[J]. 中国临床神经科学,2013,(1):70-74.
Clinical and pathological characteristic of 84 patients with myotonic dystrophy
CHEN Xi,PU Chuanqiang,ZHANG Yutong,et al.
(Department of Neurology,General Hospital of PLA,Beijing 100853,China)
Objective To explore the clinical features and muscular pathological characteristics of muscle in patients with myotonic dystrophy. Methods Eighty-four patients definitely diagnosed as myotonic dystrophy by clinical and electromyographic examination during the period of 2003 to 2015 were studied. Open biopsy were performed to obtain the skeletal muscle samples. The samples were stained by histochemical staining and observed under light microscope. Results Among 84 cases,71 had myotonia,and 57 had muscle weakness. 39 patients suffered from muscle wasting. 11 cases were diagnosed with cataract. Under the light microscope,myopathic changes were found in 79 patients. 62 of the 79 biopsies showed fiber size variation and 78 biopsies had muscle fiber atrophy. 59 of the 79 cases presented internalized nuclei. The sarcoplasmic masses were shown in only 29 cases. There were significant differences in age between the groups with myotonia and without myotonia(P=0.008). Conclusion The core features in DM are muscle weakness,myotonia,muscle wasting,and cataract. Central nucleation and sarcoplasmic masses are important pathological characteristics in DM.
Myotonic dystrophy; Muscle biopsy; Pathology; Sarcoplasmic masses
1003-2754(2016)03-0200-03
2016-01-15;
2016- 02-15
(1.解放军总医院神经内科,北京 100853;2.南开大学医学院,天津 300071;3.山西医科大学第一附属医院神经内科,山西 太原 030003)
蒲传强,E-mail:pucq30128@sina.cn
R746.2
A
