Role of stromal cell-derived factor 1α pathway in bone metastatic prostate cancer
2016-12-13NishaGuptaDanDuda
Nisha Gupta, Dan G. Duda
Steele Laboratories for Tumor Biology, Department of Radiation Oncology, Massachusetts General Hospital Research Institute, Harvard Medical School, 100 Blossom Street, Boston, MA 02114, USA.
Role of stromal cell-derived factor 1α pathway in bone metastatic prostate cancer
Nisha Gupta, Dan G. Duda✉
Steele Laboratories for Tumor Biology, Department of Radiation Oncology, Massachusetts General Hospital Research Institute, Harvard Medical School, 100 Blossom Street, Boston, MA 02114, USA.
Metastatic prostate cancer is one of the leading causes of cancer-related death in men. The primary site of metastasis from prostate cancers is the bone. During the last decade, multiple studies have pointed to the role of the stromal cell-derived factor 1 alpha (SDF1α)/CXCR4 axis in the metastatic spread of the disease, but the mechanisms that underlie this effect are still incompletely understood. In this review, we summarize the current understanding of the role of the SDF1α/CXCR4 pathway in bone metastatic prostate cancer. We also discuss the therapeutic potential of disrupting the interaction between prostate tumor cells and bone environment with focus on the SDF1α pathway.
SDF1α, CXCR4, bone metastasis, prostate cancer
Introduction
Prostate cancer is the second most common cancer among men and the second leading cause of cancerrelated death in men in the United States. Most prostate cancers (93%) are found when the disease is confined to the prostate and nearby organs, is indolent, and has a good prognosis. However, the 5-year survival rate sharply declines from 90% for localized prostate cancer to 28% for metastatic prostate cancer[1]. The skeletal bones are the preferential sites of metastasis from prostate cancer[2]. In fact, homing of metastatic prostate cancer cells to bone tissue is associated with the presence and activity of osteoblast lineage cells[3-6]. However, the precise mechanism leading to prostate cancer bone metastasis remains poorly understood. The process of metastasis is known to be the result of several necessary sequential steps - including the survival of tumor cells at distant locations and adaption to the foreign microenvironment, thereby facilitating cell proliferation and the formation of a metastatic lesion. The aggressive and highly metastatic capacity makes the treatment of advanced prostate cancer a major challenge[7]. The therapeutic options currently available (local radiation, cytotoxics, vaccine therapy, and hormonal therapies) are palliative and cannot control disease progression. Despite the clinical significance of bone metastatic prostate cancer, we only know little about the molecular mechanisms underlying the progression of this disease.
Role of chemokines in metastatic prostate cancer: the SDF1α pathway
Chemokines are a family of small (8 to 12 kDa) peptides that function as chemo-attractant cytokines that mediate and regulate cell activation, differentiation and trafficking. Chemokines are known to interact with a superfamily of 20 C-C or C-X-C trans-membrane domainheterotrimeric G protein-coupled receptors (GPCR)[8]. The stromal-derived factor 1 alpha (SDF1α), also referred to as CXCL12, binds and initiates signaling through its receptors C-X-C chemokine receptor type 4 (CXCR4) and C-X-C chemokine receptor type 7 (CXCR7)[9,10]. The SDF1α/CXCR4 signaling has been recognized as a critical pathway for the homing and tissue retention of hematopoietic progenitor/stem cells in the bone marrow microenvironment[11]. Several studies have shown that CXCR4 plays a crucial and pleiotropic role in malignant tumor progression, including prostate cancer, particularly in the metastatic spread of the disease. High levels of the chemokine receptor CXCR4 induce a more aggressive phenotype in prostate cancer cells[12,13]. Interestingly, the bone environment - in which SDF1α is particularly highly expressed - is also the most common metastatic site of prostate cancer. Moreover, metastatic prostate cancer cells localized in the bone metastatic lesions express higher SDF1α/CXCR4 levels relative to the cells present in primary tumors and lymph node metastatic lesions[6,14].
These findings suggest that the activation of the SDF1α/ CXCR4 pathway may play a pivotal role in prostate cancer bone metastases. This review focuses primarily on the SDF1α/CXCR4 axis regulation, on the pre-clinical observations made in bone metastatic prostate cancer metastases, and their implication for development of more effective treatment strategies in the future.
The SDF1α/CXCR4 axis: Role in bone metastatic prostate cancer
SDF1α signaling can be activated via CXCR4 in prostate cancer cells driven by the loss of phosphatase and tensin homolog (PTEN) and subsequent activation of PI3K/Akt pathway. Akt1-associated SDF1α/CXCR4 signaling can promote prostate tumor growth[15]. Moreover, silencing of CXCR4 can lead to a significant down- regulation in the secretion of vascular endothelial growth factor (VEGF) and matrix metalloproteinase 9 (MMP-9), to a delay in primary tumor growth and to inhibition of the incidence of prostate cancer bone metastases[16].
SDF1α is also produced by the bone marrow stromal cells of mesenchymal origin, including osteoblasts, and by vascular endothelial cells[5,17,18]. SDF1a transiently regulates the number and affinity of αvβ3receptors by prostate cancer cells, and increases the expression of the β3subunit to enhance their metastatic behavior by increasing adhesiveness and invasiveness in bone marrow[19]. Additionally, SDF1α induces the expression of CD164 and blockade of CD164 in prostate cancer cell lines decreases the ability of these cells to adhere to human bone marrow endothelial cells[20]. This suggests that activation and increased expression of CD164 and αvβ3may be important in the metastatic spread of prostate cancer cells to the skeleton. In addition, inhibition of CXCR4 activity alters the homing of quiescent prostate cancer cells to bone. These cells have more potential to form bone metastases than rapidly proliferating prostate cancer cells. Higher levels of CXCR4 are associated with mitotic dormancy that facilitates tumor cell colonization of the bone marrow in prostate cancer[21-23]. These dormant or slow-cycling disseminated prostate cancer cells in bone marrow are more resistant to conventional therapies. It was shown that these cells predominantly express transforming growth factor-beta 2 (TGF-β2) to maintain SDF1α/CXCR4 overexpression[24].
Several studies showed that metastatic prostate cancers recruit mesenchymal stem cells, which are converted into cancer-associated fibroblasts and facilitate metastasis. In prostate tumors, CXCL16, a ligand for CXCR6, stimulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts, which secrete higher levels of SDF1α. SDF1α binds to CXCR4 on prostate cancer cells and induces an epithelial-to-mesenchymal transition (EMT)[25]. Inhibition of the SDF1α/CXCR4 axis leads to suppression of the bone marrow mesenchymal stem cell-induced prostate cancer stem cell population increase, and to downregulation of the expression of MMP-9, ZEB-1, CD133 and CXCR4[26]. Accumulating evidence suggests that activated hypoxia-inducible factors, HIF-1α and HIF-2α, may induce the expression of EMT program-associated molecules, including Snail, Twist, CXCR4 and angiogenic factors such as VEGF[27]. Interestingly, in an osteosarcoma study, hypoxia-induced CXCR4 expression persisted even after cultured cells were returned to normoxic conditions, suggesting more persistent phenotypic changes[28]. In summary, activation of the SDF1α/CXCR4 pathway may play a critical role via multiple mechanisms: by promoting prostate tumor growth, homing of prostate cancer cells to bone, and facilitating interactions among cancer cells and bone environment (Fig. 1).
The SDF1α/CXCR4 axis in bone metastasis: Beyond prostate cancer
As summarized above, a large amount of data strongly suggest a key role of the SDF1α/CXCR4 pathway in bone metastases from prostate cancer patients. However, the evidence for the involvement of this pathway in other frequent bone metastatic cancers, such as breast cancer, is very limited. Same as in prostate cancer, high levels of CXCR4 expression in breast cancer are associated with early distant and bone metastases. In various bone metastatic breast models, SDF1α expression was regulated by cytokines such as interleukin (IL)-17A, IL-6, macrophage colony-stimulating factor (M-CSF), liver-enriched inhibitory protein (LIP), neuregulin 1 (NRG1), SMAD4, HIF-1, BACH1 and MMP-1[29-32]. Since tumor-stroma interactionsinvolving the SDF1α/CXCR4 are likely to be equally important in these settings, further understanding in this area may have important implications for understanding bone metastasis in general. Moreover, this improved understanding could potentially allow the development of novel strategies to reduce morbidity and mortality in bone metastatic cancer patients.
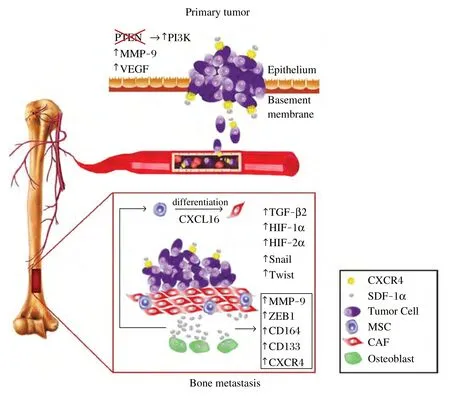
Fig. 1 Pleiotropic role of SDF1α/CXCR4 Axis in Prostate Cancer Bone Metastasis. SDF-1α/CXCR4 axis activation in prostate cancer cells could be driven by oncogenic events (e.g., the loss of PTEN and subsequent activation of PI3K/Akt pathway), or by microenvironmental factors (e.g., hypoxia, fibrosis, inflammation). When CXCR4 is inhibited, it could lead to inhibition of VEGF and MMP-9 expression, a delay in tumor growth and reduction in metastasis. SDF1α can induce CD164 expression in prostate cancer cells and there by increase their ability to adhere to human bone marrow endothelial cells. In bone metastases, CXCL16 (a ligand for CXCR6) can stimulate the differentiation of mesenchymal stem/stromal cells (MSCs) into cancer-associated fibroblasts (CAFs), which secrete high levels of SDF1α. SDF1α could also be locally produced by osteoblasts. SDF1α and CXCR4 overexpression could also be stimulated by TGF-β2, which is expressed by dormant disseminated prostate cancer cells in bone marrow. Finally, hypoxiainduced factor (HIF)-1a and HIF-2a may induce the expression of epithelial-to mesenchymal-transition (EMT)-associated molecules snail and twist. In turn, inhibition of SDF-1α could lead to downregulation of the expression of MMP-9, ZEB-1, CD133 and CXCR4 in bone metastatic prostate tumors. Figure courtesy of Rekha Gupta and Priscilla R.C. Jainandunsing.
SDFlα and bone metastatic prostate cancer therapy: Future perspective
The increasing evidence that SDF1α/CXCR4 signaling may contribute to prostate cancer progression and relapse warrants studies of SDF1α/CXCR4 inhibition to target both primary or metastatic lesions. The most studied SDF1α/CXCR4 inhibitor is the CXCR4 antagonist AMD3100, an FDA-approved drug known as plerixafor or Mozobil®and used for hematopoietic stem cell mobilization by bolus injection. AMD3100 has been shown to inhibit primary tumor growth and decrease the incidence of metastases in multiple animal models[33-37]. However, the safety of chronic treatment with a chemokine receptor antagonist remains to be confirmed in patients, given the obvious concerns related to hematologic toxicities.
An alternative approach would be to inhibit the expression of SDF1α/CXCR4 in primary and metastatic prostate cancers, without directly inhibiting CXCR4 signaling. In this respect, it is critical to identify the mechanism by which CXCR4 expression is activated in prostate cancer bone metastases, in particular by hypoxia induced by tumor growth or by therapeutic interventions[38,39]. Moreover, multiple molecular pathways are likely required to work in concert with the SDF1α/CXCR4 pathway for the homing of metastatic prostate cancer cells to the bone. Many of the interactions among prostate cancer cells and the bone environment remain to be characterized. Finally, sustained responses in this disease, same as in other cancers, will most likely require the development ofefficacious immunotherapies. The currently available vaccine has only limited efficacy, and checkpoint inhibitors have failed so far to show promising efficacy in advanced prostate cancer patients. Combining CXCR4 inhibition with checkpoint blockade, with or without radiation therapy, may be a potential strategy to achieve synergy[33]. Once this improved understanding is achieved, preclinical and clinical studies should translate this knowledge into real benefits for bone metastatic prostate cancer patients.
Acknowledgments
DGD’s research is supported by NIH grants R01-CA159258 and P01-CA080124 and by American Cancer Society Grant 120733-RSG-11-073-01-TBG.
References
[1] American Cancer Society American Cancer Society:Cancer Facts and Figures 2015.
[2] Jimenez-Andrade JM, Mantyh WG, Bloom AP, et al. Bone cancer pain[J]. Ann N Y Acad Sci, 2010,1198:173-181.
[3] Hart CA, Brown M, Bagley S, et al. Invasive characteristics of human prostatic epithelial cells: understanding the metastatic process[J]. Br J Cancer, 2005,92:503-512.
[4] Wang N, Docherty FE, Brown HK, et al. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models[J]. J Bone Miner Res, 2014,29(12):2688-2696.
[5] Sun YX, Schneider A, Jung Y, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo[J]. J Bone Miner Res, 2005,20(2):318-329.
[6] Miwa S, Mizokami A, Keller ET, et al. The bisphosphonate YM529 inhibits osteolytic and osteoblastic changes and CXCR-4-induced invasion in prostate cancer[J]. Cancer Res, 2005,65(19):8818-8825.
[7] Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited[J]. Nat Rev Cancer, 2003, 3(6):453-458.
[8] Duda DG, Kozin SV, Kirkpatrick ND, et al. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies?[J] Clin Cancer Res, 2011,17(8):2074-2080.
[9] Sun X, Cheng G, Hao M, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression[J]. Cancer Metastasis Rev, 2010,29(4):709-722.
[10] Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis[J]. Nature, 2001,410(6824):50-56.
[11] Hirbe AC, Morgan EA, Weilbaecher KN. The CXCR4/ SDF-1 chemokine axis: a potential therapeutic target for bone metastases?[J] Curr Pharm Des, 2010,16(11):1284-1290.
[12] Saylor PJ, Kozak KR, Smith MR, et al. Changes in biomarkers of inflammation and angiogenesis during androgen deprivation therapy for prostate cancer[J]. Oncologist, 2012,17(2):212-219.
[13] Darash-Yahana M, Pikarsky E, Abramovitch R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis[J]. FASEB , 2004,18(11): 1240-2.
[14] Domanska UM, Timmer-Bosscha H, Nagengast WB, et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy[J]. Neoplasia, 2012,14(8): 709-718.
[15] Conley-LaComb MK, Saliganan A, Kandagatla P, et al. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling[J]. Mol Cancer, 2013,12(1):85.
[16] Wang Q, Diao X, Sun J, et al. Regulation of VEGF, MMP-9 and metastasis by CXCR4 in a prostate cancer cell line[J]. Cell Biol Int, 2011,35(9):897-904.
[17] Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor- 1/CXCR4 pathway in prostate cancer metastasis to bone[J]. Cancer Res, 2002,62(6):1832-1837.
[18] Al Nakouzi N, Bawa O, Le Pape A, et al. The IGR-CaP1 xenograft model recapitulates mixed osteolytic/blastic bone lesions observed in metastatic prostate cancer[J]. Neoplasia, 2012,14:376-387.
[19] Sun YX, Fang M, Wang J, et al. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells[J]. Prostate, 2007, 67(5):61-73.
[20] Havens AM, Jung Y, Sun YX, et al. The role of sialomucin CD164 (MGC-24v or endolyn) in prostate cancer metastasis[J]. BMC Cancer, 2006,6:195.
[21] Wang N, Docherty F, Brown HK, et al. Mitotic quiescence, but not unique “sternness,” marks the phenotype of bone metastasis-initiating cells in prostate cancer[J]. FASEB J, 2015,29(8):3141-3150.
[22] Xing Y, Liu M, Du Y, et al. Tumor cell-specific blockade of CXCR4/SDF-1 interactions in prostate cancer cells by hTERT promoter induced CXCR4 knockdown: A possible metastasis preventing and minimizing approach[J]. Cancer Biol Ther, 2008,7(11):1839-1848.
[23] Du YF, Shi Y, Xing YF, et al. Establishment of CXCR4-small interfering RNA retrovirus vector driven by human prostate-specific antigen promoter and its biological effects on prostate cancer in vitro and in vivo[J]. J Cancer Res Clin Oncol, 2008,134(11):1255-1264.
[24] Nakamura T, Shinriki S, Jono H, et al. Intrinsic TGF-beta2-triggered SDF-1-CXCR4 signaling axis is crucial for drug resistance and a slow-cycling state in bone marrow-disseminated tumor cells[J]. Oncotarget, 2015,6(2):1008-1019.
[25] Jung Y, Kim JK, Shiozawa Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis[J]. Nat Commun, 2013,4:1795.
[26] Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling[J]. Oncogene, 2014,33(21):2768-78.
[27] Mimeault M, Batra SK. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells[J]. J Cell Mol Med, 2013,17(1):30-54.
[28] Guan G, Zhang Y, Lu Y, et al. The HIF-1alpha/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells[J]. Cancer Lett, 2015,357(1):254-264.
[29] Hung CS, Su HY, Liang HH, et al. High-level expression of CXCR4 in breast cancer is associated with early distant and bone metastases[J]. Tumour Biol, 2014,35(2):1581- 1588.
[30] Liang Y, Wu H, Lei R, et al. Transcriptional network analysis identifies BACH1 as a master regulator of breast cancer bone metastasis[J]. J Biol Chem, 2012,287(40):33533-33544.
[31] Park BH, Kook S, Lee S, et al. An isoform of C/EBPbeta, LIP, regulates expression of the chemokine receptor CXCR4 and modulates breast cancer cell migration[J]. J Biol Chem, 2013,288(40):28656-28667.
[32] Roy LD, Sahraei M, Schettini JL, et al. Systemic neutralization of IL-17A significantly reduces breast cancer associated metastasis in arthritic mice by reducing CXCL12/ SDF-1 expression in the metastatic niches[J]. BMC Cancer, 2014,14:225.
[33] Chen Y, Ramjiawan RR, Reiberger T, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib- treated hepatocellular carcinoma in mice[J]. Hepatology, 2015,61(5):1591-1602.
[34] Kozin SV, Kamoun WS, Huang Y, et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation[J]. Cancer Res, 2010,70:5679-5685.
[35] Liao YX, Fu ZZ, Zhou CH, et al. AMD3100 reduces CXCR4-mediated survival and metastasis of osteosarcoma by inhibiting JNK and Akt, but not p38 or Erk1/2, pathways in in vitro and mouse experiments[J]. Oncol Rep, 2015,34(1):33-42.
[36] Rabenstein M, Hucklenbroich J, Willuweit A, et al. Osteopontin mediates survival, proliferation and migration of neural stem cells through the chemokine receptor CXCR4[J]. Stem Cell Res Ther, 2015,6:99.
[37] Hiratsuka S, Duda DG, Huang Y, et al. C-X-C receptor type 4 promotes metastasis by activating p38 mitogenactivated protein kinase in myeloid differentiation antigen (Gr-1)-positive cells[J]. Proc Natl Acad Sci USA, 2011, 108(1):302-7.
[38] Chen Y, Huang Y, Reiberger T, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromalderived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice[J]. Hepatology, 2014,59(4): 1435-1447.
[39] Jain RK. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia[J]. Cancer Cell, 2014, 26(5):605-622.
✉ Dan G. Duda, DMD, PhD, Steele Laboratories for Tumor Biology, Department of Radiation Oncology, Massachusetts General Hospital Research Institute, 100 Blossom Street, Boston, MA 02114, USA. E-mail: gduda@partners.org.
8 August 2015, Accepted 10 October 2015, Epub 2 November 2015
R737.25, Document code: A
The author reported no conflict of interests.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Lipid transport to avian oocytes and to the developing embryo
- A pilot exome-wide association study of age-related cataract in Koreans
- Dynamic monitoring of menopause hormone therapy and defining the cut-off value of endometrial thickness during uterine bleeding
- Polycystic ovary syndrome patients with high BMl tend to have functional disorders of androgen excess: a prospective study
- Effect of vitamin D3 on production of progesterone in porcine granulosa cells by regulation of steroidogenic enzymes
- Human lgG Fc promotes expression, secretion and immunogenicity of enterovirus 71 VP1 protein