Sulindac sulfide selectively increases sensitivity of ABCC1 expressing tumor cells to doxorubicin and glutathione depletion
2016-12-13JasonWhittAdamKeetonBernardGaryLarrySklarKamleshSodaniZheShengChenGaryPiazza
Jason D. Whitt, Adam B. Keeton, Bernard D. Gary, Larry A. Sklar, Kamlesh Sodani, Zhe-Sheng Chen, Gary A. Piazza,✉
1Department of Biochemistry, University of Mississippi Medical Center Cancer Institute, Jackson, MS 39216, USA;
2Drug Discovery Research Center, Mitchell Cancer Institute, University of South Alabama, Mobile, AL 36604, USA;
3ADT Pharmaceuticals Inc., Orange Beach, AL 36561, USA;
4Department of Pathology, The University of New Mexico, Albuquerque, NM 87131, USA;
5Department of Pharmaceutical Sciences, St. John's University, New York, NY 11439, USA.
Sulindac sulfide selectively increases sensitivity of ABCC1 expressing tumor cells to doxorubicin and glutathione depletion
Jason D. Whitt1, Adam B. Keeton2,3, Bernard D. Gary2, Larry A. Sklar4, Kamlesh Sodani5, Zhe-Sheng Chen5, Gary A. Piazza2,3,✉
1Department of Biochemistry, University of Mississippi Medical Center Cancer Institute, Jackson, MS 39216, USA;
2Drug Discovery Research Center, Mitchell Cancer Institute, University of South Alabama, Mobile, AL 36604, USA;
3ADT Pharmaceuticals Inc., Orange Beach, AL 36561, USA;
4Department of Pathology, The University of New Mexico, Albuquerque, NM 87131, USA;
5Department of Pharmaceutical Sciences, St. John's University, New York, NY 11439, USA.
ATP-binding cassette (ABC) transporters ABCC1 (MRP1), ABCB1 (P-gp), and ABCG2 (BCRP) contribute to chemotherapy failure. The primary goals of this study were to characterize the efficacy and mechanism of the nonsteroidal anti-inflammatory drug (NSAID), sulindac sulfide, to reverse ABCC1 mediated resistance to chemotherapeutic drugs and to determine if sulindac sulfide can influence sensitivity to chemotherapeutic drugs independently of drug efflux. Cytotoxicity assays were performed to measure resistance of ABC-expressing cell lines to doxorubicin and other chemotherapeutic drugs. NSAIDs were tested for the ability to restore sensitivity to resistance selected tumor cell lines, as well as a large panel of standard tumor cell lines. Other experiments characterized the mechanism by which sulindac sulfide inhibits ABCC1 substrate and co-substrate (GSH) transport in isolated membrane vesicles and intact cells. Selective reversal of multi-drug resistance (MDR), decreased efflux of doxorubicin, and fluorescent substrates were demonstrated by sulindac sulfide and a related NSAID, indomethacin, in resistance selected and engineered cell lines expressing ABCC1, but not ABCB1 or ABCG2. Sulindac sulfide also inhibited transport of leukotriene C4into membrane vesicles. Sulindac sulfide enhanced the sensitivity to doxorubicin in 24 of 47 tumor cell lines, including all melanoma lines tested (7-7). Sulindac sulfide also decreased intracellular GSH in ABCC1 expressing cells, while the glutathione synthesis inhibitor, BSO, selectively increased sensitivity to sulindac sulfide induced cytotoxicity. Sulindac sulfide potently and selectively reverses ABCC1-mediated MDR at clinically achievable concentrations. ABCC1 expressing tumors may be highly sensitive to the direct cytotoxicity of sulindac sulfide, and in combination with chemotherapeutic drugs that induce oxidative stress.
multi-drug resistance, doxorubicin, sulindac, MRP1, glutathione
Introduction
Multi-drug resistance (MDR) is a major clinical obstacle that limits the efficacy of many cancer chemotherapeutic drugs. Tumors that progress following chemotherapy often contain populations of cells that display the MDR phenotype, which contributes to the recurrence of many types of tumors following chemotherapy. An important factor that contributes to MDR is the expression of certain ATP-dependent membrane transport proteins that cause the efflux of a number of cancer chemotherapeutic drugs, thereby reducing intracellular concentrations to limit their anti-proliferative and pro-apoptotic activity[1]. The cancer chemotherapeutic drugs most frequently affected by increased expression of transport proteins include taxanes (paclitaxel, docetaxel), vinca alkaloids (vinorelbine, vincristine, and vinblastine), anthracyclines (doxorubicin, daunorubicin, epirubicin), epipodophyllotoxins (etoposide), camptothecins (irinothecan, topotecan), dactinomycin, and mitomycin C[2].
Two of the most well studied transport proteins that contribute to drug resistance are the permeability glycoprotein (P-gp or ABCB1) that was discovered in 1976[3]and the multidrug resistance protein (MRP1 or ABCC1) that was discovered in 1992[4]. These transporters belong to a larger family of proteins referred to as the ATP-binding cassette (ABC) family, of which there are currently 48 members. Functionally, all ABC proteins are ATPases that use energy from ATP hydrolysis to transport their substrates across cell membranes. ABCB1 is a 170 kD phospho-glycoprotein encoded by the ABCB1 gene[5], while ABCC1 is a 190 kD polypeptide encoded by the ABCC1 gene[4]. Although there is a relatively small degree of sequence homology between ABCB1 and the ABCC family[6], these proteins share the ability to transport a number of commonly used chemotherapeutic drugs such as the anthracyclines and vinca alkaloids[1]. In general, ABCB1 shows preferential binding to basic hydrophobic compounds, while ABCC1 transports mainly anionic hydrophobic compounds[7]. Additional ABC proteins may also be important to MDR, for example the recently characterized breast cancer resistance protein (BCRP, ABCG2)[8], but less is known about their role in chemoresistance or substrate structural requirements.
The first generation of ABC transport inhibitors that targeted ABCB1 were non- selective and displayed low potency, leading to unacceptable toxicity. A number of newer drugs have been identified that inhibit ABCB1 with greater potency and selectivity, but also failed because these agents were found to alter the pharmacokinetic properties of many chemotherapeutic drugs[9-10]. This is generally attributed to the expression of ABCB1 in normal epithelial cells of the colon, kidney, and liver, which caused unpredictable effects on the absorption and excretion of many chemotherapeutic drugs, necessitating counterproductive dose reduction[11-12]. However, a potentially important difference between ABCB1 and ABCC1 is the role the former has in protecting normal tissues from xenobiotics. For example, ABCB1 is localized on the apical surface of normal epithelial cells of the colon, liver, and kidney and can influence the metabolism and elimination of chemotherapeutic drugs. In contrast, ABCC1 is usually localized to the basolateral surface of polarized cells except for brain capillary endothelial cells[13]. As such, it is possible that ABCC1 inhibitors may be less likely to interfere with the absorption and elimination of chemotherapeutic drugs to the same extent as ABCB1 inhibitors.
Previous reports have demonstrated the ability of cer tain nonsteroidal anti-inflammatory drugs (NSAIDs) to increase the sensitivity of ABCC1 overexpressing cells to chemotherapeutic drug substrates. For example, Duffy and colleagues performed an extensive series of in vitro experiments to evaluate the ability of vari ous NSAIDs to increase the sensitivity of ABCC1 expressing tumor cell lines to chemotherapeutic drugs[14]. These investigators concluded that the effect was independent of the cyclooxygenase- inhibitory activity of the NSAIDs, although the exact mechanism of action is not known. Interestingly, the effect was not observed in cell lines overexpressing ABCB1 but was only noted in lines that displayed ABCC1 overexpression, which suggests a direct inhibition of the protein and is consistent with selectivity of ABCC1 to transport anionic hydrophobic compounds such as NSAIDs[7,15]. There is also in vivo evidence showing that the NSAID sulindac can increase the anticancer efficacy of epirubicin, a known ABCC1 substrate[16-17]. In addition, a clinical trial demonstrated that sulindac did not interfere with the absorption or excretion of epirubicin, which is consistent with the feasibility of inhibiting transport in ABCC1 expressing cells without interfering with pharmacokinetics[18]. Aside from the direct antineoplastic activity of sulindac[19-20], there may be advantages of combining with conventional chemotherapy to prevent tumor recurrence and the emergence of drug resistant tumor cells. Here we show that the predominant metabolite of sulindac, sulindac sulfide, can potently and selectively enhance the sensitivity of ABCC1 expressing cells to chemotherapeutic drugs and we further investigated the mechanism and selectivity of this interaction.
Materials and methods
Drugs and reagents
Sulindac sulfide, indomethacin and doxorubicin were purchased from Sigma-Aldrich (St. Louis, MO, USA). ABCC1 antibody (QCRL-1, monoclonal) was purchased from Alexis Biochemicals (San Diego, CA, USA). ABCB1 antibody was purchased from Covance (Princeton, NJ, USA). Secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). All other reagents were purchased from Sigma-Aldrich unless otherwise stated.
Cell culture
Human NCI-H69 (H69), H69AR, MES- SA and MES-SA/DX5 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). MES- SA and MES-SA/DX5 cells were grown in McCoy's medium containing 10% FBS under standard cell culture conditions. Jurkat and SupT1 cells overexpressing either ABCC1 or ABCB1 were generated as previously described[21]. The human epidermoid KB carcinoma cells, KB-3-1, were propagated in Dulbecco's modified Eagle's medium with 10% FBS under 5% CO2at 37uC. The ABCC1-overexpressing cell line KB-CV60 was cloned from KB-3-1 cells and maintained in medium containing 1 mg/mL cepharanthine and 60 ng/mL vincristine. A SupT1-vincristine (Vin) drug-resistant cell line that selectively over-expresses ABCC1 has been previously characterized[22]. Ovarian Ig- MXP3 (ABCG2) and its parental Igrov1-sensitive cells were kindly provided by Dr. D. Ross (Department of Medicine, University of Maryland Greenebaum Cancer Center, Baltimore, MD). The large panel of 47 tumor cell lines were seeded and incubated under conditions as previously established by the NCI Developmental Therapeutics Program[23-24].
Cytotoxicity assays
For H69/H69AR and MES-SA/DX5 growth assays, the growth inhibitory activity of doxorubicin, sulindac sulfide, and indomethacin was determined by measurement of ATP levels, an indicator of viable cell number (Cell Titer Glo assay, Promega). For MDR reversal experiments, cells were seeded in tissue culture microtiter 96-well plates at a density of 5,000 cells/well and incubated 16 hours prior to treatment. Cells were treated with 5 μmol/L sulindac sulfide or 10 μmol/L indomethacin for 4 hours prior to addition of doxorubicin. Once treatment was complete, cells were incubated at 37oC for 72 hours for dose response experiments. For the 47 cell line panel, quadruplicate samples of each cell line were treated with 8 concentrations of Dox in the presence or absence of 5 μmol/L sulindac sulfide in a single experiment. Cell Titer Glo luminescence assays were performed according to the manufacturer's specifications using a Perkin Elmer Victor3V multi-label microplate reader. For Jurkat cells expressing either ABCC1 or ABCB1, direct compound toxicity and reversal of chemoresistance were determined as previously described[21]. In brief, a 3-log dose range of either sulindac or cyclosporine A was added to cells in the presence of 150 nmol/L vincristine or daunorubicin. On day 7, cell viability was determined using a hemacytometer and trypan blue staining. Dose response curves of cells treated with sulindac sulfide or cyclosporine A with or without the chemotherapeutic agent present were compared using GraphPad Prism software.
Glutathione assay
Cells were plated at a density of 2,500 cells per well in 96-well plates and incubated overnight at 37oC and 5% CO2.Cells were incubated 18 hours in the presence of drug or drug combinations. At the end of the incubation period, glutathione levels were measured using the GSH-Glo kit (Promega) according to the manufacturer's instructions.
LTC4transport assay
Membrane vesicles (20 mg) were prepared from KB-3-1 and KB-CV60 cells as described previously[25]. For inhibition experiments, the standard incubation medium contained membrane vesicles (25 mg of protein), 137 nmol/L3H-LTC4, 0.25 mol/L sucrose, 10 μmol/L Tris-HCl (pH 7.4), 10 μmol/L MgCl2, 2 μmol/L ATP, 10 μmol/ L phosphocreatine and 100 mg/mL creatine phosphokinase with or without unlabeled LTC4in a final volume of 50 mL. Reactions were carried out at 37uC and stopped with 3 mL of ice-cold stop solution containing 0.25 mol/L sucrose, 100 μmol/L NaCl, and 10 μmol/L Tris-HCl (pH 7.4). Samples were passed through 0.22 mm Dura pore membrane filters (Millipore, Bedford, MA, USA) under vacuum. The filters were washed three times with 3 mL of ice-cold stop solution and dried at room temperature for 30 minutes. Incorporated radioactivity was measured by the use of liquid scintillation counter. In control experiments, ATP was replaced by an equal concentration of 59-AMP. Rates of net ATP-dependent transport were determined by subtracting the values obtained in the presence of 4 μmol/L AMP from those obtained in the presence of 4 μmol/L ATP.
Immunoblotting assays
H69AR and MES-SA/DX5 cells were lysed using SDS lysis buffer containing 1% SDS, 10 mmol/L Tris pH 7.5, 7.5 ug/mL aprotonin, 5 mmol/L benzamidine, 5 mmol/ L PMSF, 50 mmol/L NaF and 1.25 mmol/L NaVaO4. Whole cell lysates were separated by SDSPAGE and transferred to nitrocellulose membranes. After blocking with 3% BSA, membranes were incubated overnight at 4uC with antibodies directed against ABCC1 or ABCB1 and subsequently with anti-mouse secondary antibody conjugated with horseradish peroxidase. β-actin antibody (Cell Signaling Technology) was used as a protein loading control. The SuperSignal West Pico Substrate kit (ThermoScientific, Waltham,MA) was used for enhanced chemiluminescence detection.
Doxorubicin/Calcein-AM confocal imaging assay
H69AR cells were plated in coverglass bottom 96-well plates and allowed to adhere overnight. Cells were then treated overnight with MRP-1 antagonists in phenol free complete growth medium. On the assay day, cells were incubated for 2 hours with 10 μmol/L doxorubicin (Dox) or 30 minutes with 0.1 μmol/L calcein-AM and 2 μmol/L Draq5 nuclear stain. At the end of the loading period, media was aspirated and replaced with phenol free medium plus MRP-1 antagonist. Plates were immediately analyzed by high speed confocal microscopy using the Evotec Opera with a 20 × water immersion objective lens. Mean intracellular intensity of Dox or calcein-AM was determined using the Acapella image analysis software.
MRP-1 indirect immunofluorescence
Cells were fixed with 4% formaldehyde (Sigma Aldrich). Samples were incubated overnight at 4uC with anti-MRP-1 antibody followed by AlexaFluor 488 labeled anti-mouse antibody conjugate (Life Technology). Nuclei were stained with DAPI (Sigma Aldrich; 1 mg/mL) for 30 minutes at room temperature. Fluorescent images were obtained as above.
Laser scanning cytometry assay
H69AR cells were plated in cover glass bottom 96-well plates and allowed to adhere overnight. On the assay day, cells were incubated for 3.67 hours with a dilution series of sulindac sulfide followed by incubation for 20 minutes with 100 nmol/L calcein-AM. At the end of the loading period, free calcein-AM was washed away with PBS. Cellular fluorescence was analyzed using a Blueshift Isocyte laser scanning cytometer.
Statistical analysis
Concentration-response studies and IC50 values were analyzed using GraphPad Prism software. With the exception of the 47 cell-line panel, experiments were repeated three or more times to ensure reproducibility of results. Analysis of differences between means was determined using Student's T-test, or where indicated, analysis of variance (ANOVA). For samples with P,0.05, differences were considered significant.
Transporter activity assay
Cells expressing ABCB1 (JurkatDNR), ABCC1 (SupT1-Vincristine), or ABCG2 (IgMxp3) were generated as previously described[26-27]. Sulindac sulfide was added to cell suspensions to a final concentration of 50 μmol/L and incubated for 10 minutes at room temperature. Calcein-AM (250 nmol/ L) was then added and incubated for additional 15 minutes at room temperature. The fluorescent signal of the cells was evaluated in the HyperCyt flow cytometry system (IntelliCyt, Albuquerque, NM, USA) as previously described[26].
Results
Characterization of tumor cell lines expressing ABCC1 and ABCB1
Initial experiments were performed to compare the expression of ABCC1 and ABCB1 in the human H69AR small cell lung and MES-SA/DX5 uterine sarcoma tumor cell lines, which were each derived from DOX sensitive cell lines by in vitro selection with Dox[28-29]. As previously reported, H69AR cells express ABCC1[30]and MES- SA/DX5 cells express ABCB1[31-32]. As determined by Western blotting, ABCC1 was not detected in the parental cell line H69 or in MES-SA/ Dx5 cells, but was highly expressed in H69AR cells (Fig. 1A). By contrast, ABCB1 was highly abundant in MES-SA/ Dx5 cells compared with the parental MES-SA cells, but was minimally detectable in H69AR cells (Fig, 1B). ABCC1 was expressed at essentially uniformly high levels within the H69AR cells, but not detectable in MES-SA/ Dx5 cells by indirect immunofluorescence (Fig. 1C). Experiments were next performed to determine the potency of Dox to inhibit the growth of both pairs of sensitive and resistant cell lines. As shown in Fig. 1D, Dox inhibited the growth of H69 and H69AR cells with an IC50of 0.12 and 4.0 μmol/L, respectively, which reflects a 33-fold difference in sensitivity. Similarly, MES-SA cells were approximately 17 times more sensitive to Dox compared with MES-SA/Dx5 cells with IC50values of 0.21 μmol/L and 3.55 μmol/L, respectively. Moreover, the IC50values of sensitive lines are below clinically achievable blood levels of Dox (0.4-2.0 μmol/L), whereas IC50values in resistant lines are significantly greater than clinically achievable blood levels[33]. These results confirm the mechanism and extent of MDR in the ABCC1 and ABCB1 cell models used for experiments described below.
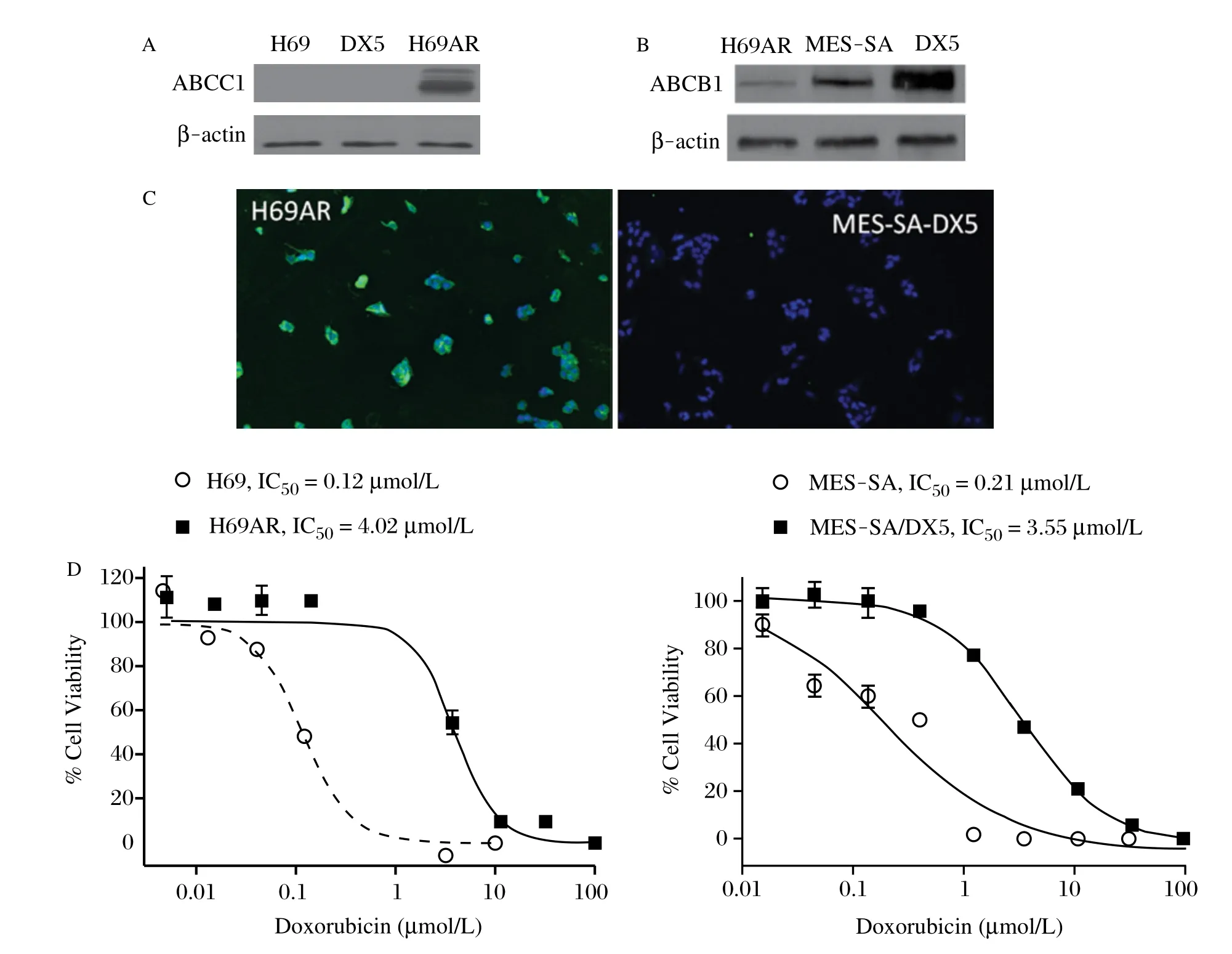
Fig. 1 Characterization of MDR cell models. A: Western blots of parental H69 cells compared to multidrug resistant H69AR and MES-SA/ DX5 uterine sarcoma cell line. B: Western blots of H69AR, MES-SA, and the drug resistant variant MES-SA/DX5 cells showing relative levels of ABCB1/P-glycoprotein expression. C: Immunofluorescent detection of ABCC1 in H69AR (left) and MES-SA/DX5 cells (right). D: MRP1 expressing (H69AR) and P-gp expressing (MES-SA/DX5) cells were treated with increasing concentrations of doxorubicin to quantitate drug resistance compared to their parental lines, H69 and MES-SA respectively.
SS and indomethacin increase sensitivity to Dox in ABCC1 overexpressing cells
ABCC1 overexpressing H69AR cells and the H69 parental line were treated with sulindac sulfide or a chemically related NSAID, indomethacin to determine their sensitivity to growth inhibition in the presence of either compound alone (Fig. 2A). Subtoxic concentrations of Dox at 25 and 500 nmol/L that correspond to their approximate IC20value to inhibit the growth of H69 and H69AR cells, respectively, were selected to distinguish between additive toxicity and ABCC1 inhibition. Both cell lines were pre-treated for 4 hours with SS or indomethacin over a concentration range of 1-500 μmol/L prior to the addition of Dox. Both sulindac sulfide and indomethacin significantly increased the sensitivity of drug resistant H69AR cells to Dox (Fig. 2B), but did not significantly affect the sensitivity of parental H69 cells to Dox (Fig. 2C). Sulindac sulfide increased cytotoxicity of 500 nmol/L Dox by 30-40% within a concentration range of 2-32 μmol/L, while indomethacin increased cytotoxicity by 20-30% within the same concentration range. The effective concentration range of sulindac sulfide and indomethacin was significantly less than the concentration range at which the drugs were cytotoxic as single agents, which suggests that the mechanism is unrelated to their knowntumor cell growth inhibitory activity[20,34]. In the case of sulindac sulfide, the effect was within the concentration range that can be achieved clinically with standard dosages of sulindac[35].

Fig. 2 Potency determination of NSAIDs. A: Growth inhibition of H69AR and parental H69 cells by sulindac sulfide or indomethacin was measured after 3 days of treatment. H69AR (B) and H69 (C) cells were treated with increasing concentrations of sulindac sulfide or indomethacin before addition of subtoxic concentrations of Dox (500 nmol/L and 25 nmol/L, respectively) to determine the concentration range over which the NSAIDs enhance sensitivity to Dox. Data is expressed as the difference between cytotoxicity of NSAID alone versus NSAID+Dox (Single factor ANOVA, P≤0.05).
To quantify the reversal of resistance by sulindac sulfide and indomethacin, we selected a single sub-toxic concentration of sulindac sulfide and indomethacin that caused MDR reversal and varied the concentration of Dox. Each drug decreased the IC50 value of Dox in H69AR cells by approximately 18-fold (Fig. 3A, left). By contrast, neither drug significantly affected the IC50value of Dox to inhibit H69 cell growth (Fig. 3A, right). Sulindac sulfide also did not significantly enhance the sensitivity of the ABCB1 expressing MES-SA/Dx5 cells to Dox (Fig. 3B, left), while a known inhibitor of ABCB1, cyclosporine A, decreased the IC50 value from 2.2 μmol/L to 0.1 μmol/L (Fig. 3B, right).
To confirm that the effects of sulindac sulfide were specific for ABCC1 rather than the process of selection of MDR variants, we utilized Jurkat cells that expressed either ABCC1 or ABCB1 by treatment with incrementally increasing doses of daunorubicin or vincristine[21,26-27]. Both ABCC1 and ABCB1 overexpressing cells were treated with increasing concentrations of sulindac sulfide and a sub-toxic dose of vincristine (150 nmol/L) to determine the ability ofthe NSAID to enhance sensitivity. Sulindac sulfide at or above 1 μmol/L reversed the ABCC1 mediated vincristine resistance (Fig. 3C). By contrast, sulindac sulfide provided no significant enhancement of vincristine cytotoxicity in Jurkat cells expressing ABCB1 (Fig. 3D). Treatment with sulindac sulfide alone demonstrated that the restoration of chemosensitivity was not due to cytotoxicity of sulindac. As demonstrated with MES-SA/DX5 cells, a known inhibitor of ABCB1, cyclosporine A, reversed the chemoresistance to vincristine in ABCB1 overexpressing cells (Fig. 3E).
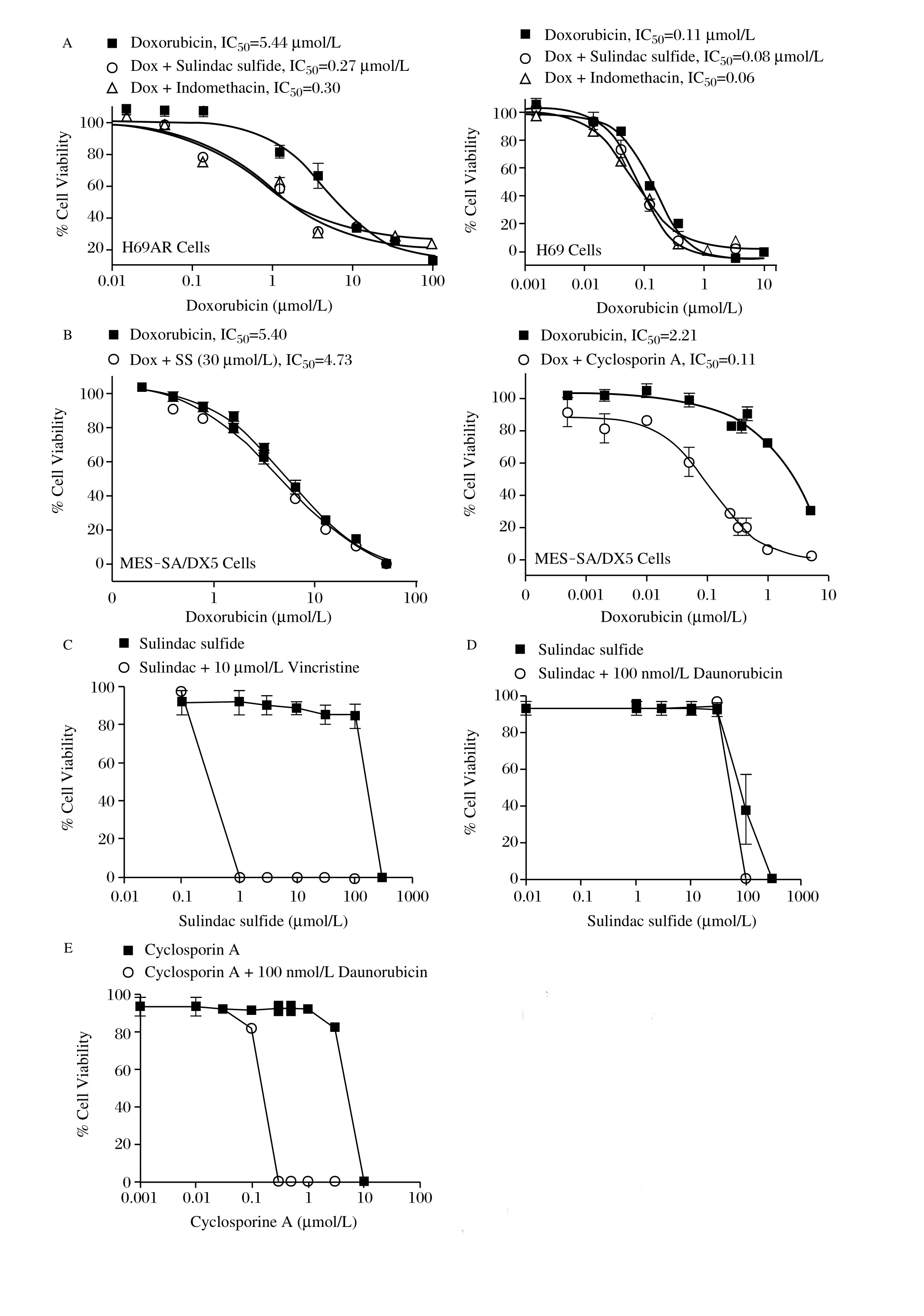
Fig. 3 Selectivity of NSAIDs for ABCC1. A: H69AR and the parental H69 cells were treated with doxorubicin in the presence of the IC20of either sulindac sulfide or indomethacin. The doses of NSAIDs used were 5 μmol/L for sulindac sulfide and 10 μmol/L for indomethacin. B: The ABCB1 expressing uterine sarcoma cell line MES-SA/DX5 was treated with doxorubicin and either 10 μmol/L sulindac sulfide or 30 μmol/L cyclosporin A. C: The ability of sulindac sulfide to sensitize cells to a structurally distinct cytotoxic agent, vincristine, was tested in Jurkat cells expressing ABCC1. Cells were treated with sulindac sulfide alone (squares) to determine the toxicity of sulindac sulfide alone in these cells. The ability of sulindac sulfide (D) or cyclosporin A (E) to sensitize cells to growth inhibition by daunorubicin was determined in Jurkat cells expressing ABCB1. Columns represent the mean of triplicate determinations while bars indicate standard deviation. *P<0.05; **P<0.01, versus the control group.
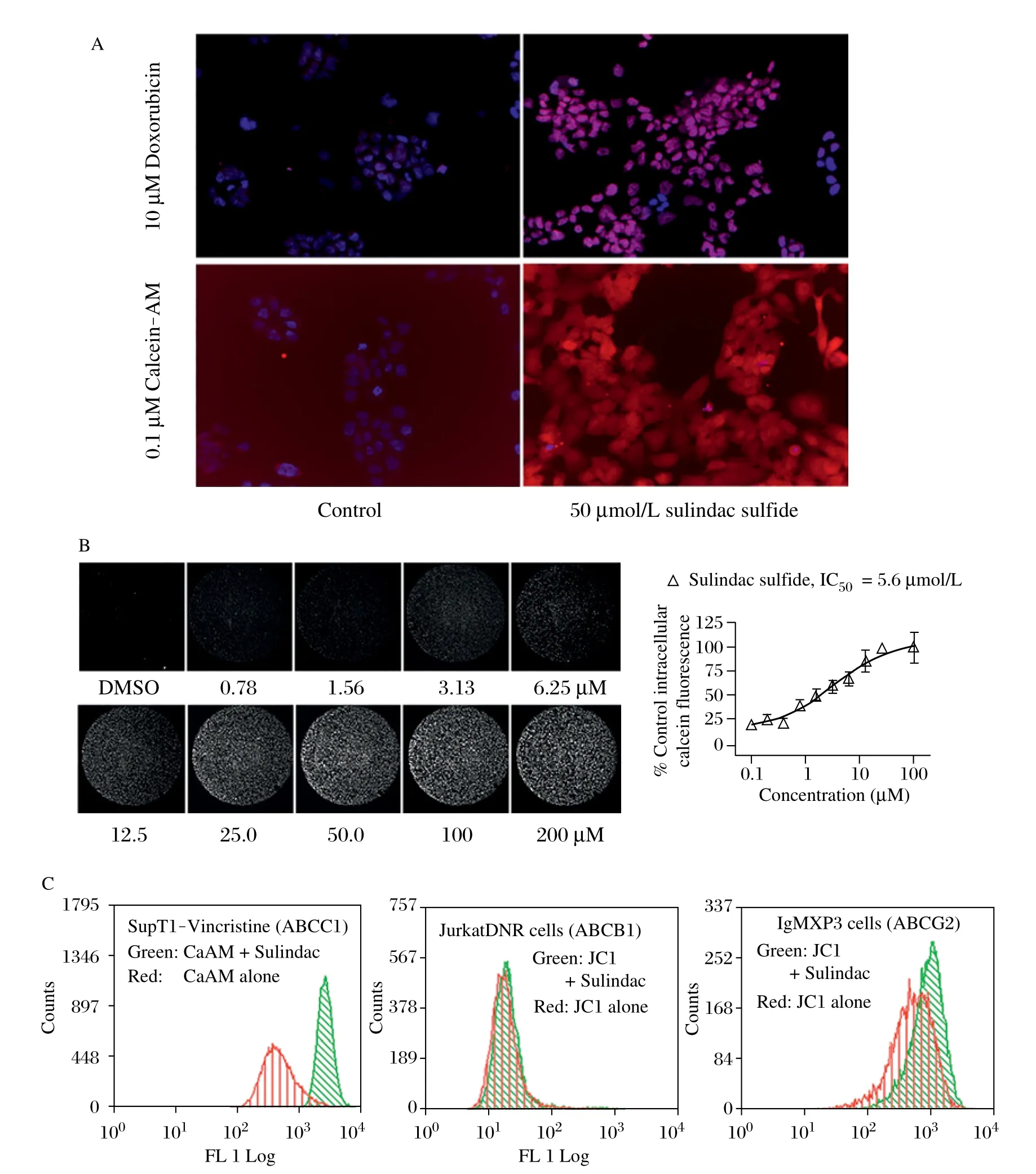
Fig. 4 Specific inhibition of ABCC1-mediated efflux by sulindac sulfide. A: Intracellular accumulation of calcein-AM or doxorubicin in H69AR cells pretreated with 50 μmol/L sulindac sulfide measured by confocal laser microscopy using 20x magnification objective lens. B: The potency of sulindac sulfide to inhibit ABCC1 mediated efflux of fluorescent calcein from H69AR cells was measured by laser scanning cytometer. Each image depicts a complete 6.35 μmol/L diameter well of a 96-well microplate. Plot represents analysis of fluorescence intensity of 4 replicate wells for each concentration. C: Fluorescent intensity of calcein-AM (ABCC1 substrate) or JC1 (ABCB1, ABCG2 substrate) in the presence of SS (50 mmol/L) was measured by flow cytometry.
Sulindac sulfide inhibits ABCC1-mediated efflux
We next performed studies to determine the mechanism by which sulindac sulfide increases sensitivity in ABCC1 expressing tumor cells. Increased intracellular autofluorescence of Dox or the fluorogenicsubstrate calcein-AM was demonstrated in H69AR cells in the presence of sulindac sulfide by confocal microscopy (Fig. 4A). Next, adherent cultures of H69AR cells were pretreated with a range of concentrations of sulindac sulfide, followed by 30-minuteincubation with calcein AM. After cells were washed to remove free calcein-AM, the fluorescence intensity of retained intracellular calcein was measured using a laser scanning fluorimeter (Fig. 4B). These studies indicated that sulindac sulfide inhibited ABCC1-mediated efflux with an IC50 value of 5.6 μmol/L and consistent with the concentration required to enhance sensitivity of H69AR cells to inhibition of growth by Dox.
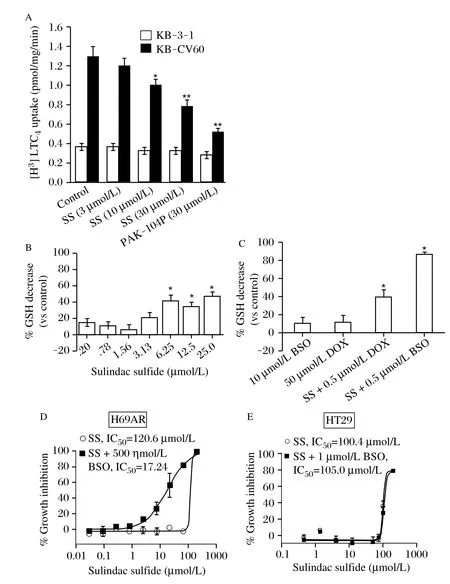
Fig. 5 LTC4 transport and glutathione depletion. A: Inhibition of ATP-dependent transport of [3H]-LTC4 into ABCC1-positive (KB-CV60) and ABCC1-negative (KB-3-1) membrane vesicles by sulindac sulfide (SS) and the non-specific inhibitor PAK-104P. B: Intracellular GSH levels in H69AR cells treated with non-cytotoxic concentrations of SS for 18 hours. (ANOVA , *P≤0.05). C: The effect on intracellular glutathione levels by SS alone and in the presence of a non-cytotoxic concentration of Dox or BSO (ANOVA, P<0.05). (Columns, mean of three experiments; bars, SEM; *, significant, P<0.05). D and E: Growth inhibition of ABCC1 expressing H69AR cells or the colon cancer cell line HT-29 by SS alone or in combination with BSO (1 μmol/L).
Population analysis of transporter activity was next evaluated using fluorescent substrates for three different ABC transporters, calcein-AM for ABCC1 and JC1 for ABCB1 and ABCG2. The distribution of fluorescence intensity of ABCC1 overexpressing cells (SupT1-Vin) was significantly increased in the presence of sulindac sulfide with the mean cellular fluorescence (MCF) increasing over 10-fold from 234 ± 32.9in untreated cells to 2445 ± 73.8 in the presence of sulindac sulfide. In contrast, sulindac sulfide treatment caused very little change in fluorescent intensity in cells overexpressing either ABCB1 or ABCG2 (Fig. 4C).

Table 1 Sulindac sulfide increases sensitivity of human tumor cell lines to doxorubicin.
Effect of sulindac sulfide on ABCC1 mediated transport of LTC4
In order to determine if the observed cellular effects of sulindac sulfide occur directly on the ABCC1 protein, the transport of the leukotriene, LTC4,was measured. LTC4is a high affinity physiological substrate of ABCC1[36]. Inside out membrane vesicles were isolated from the ABCC1 expressing clone KB-CV60. The transport of LTC4into the vesicles was measured in the presence of sulindac sulfide or the pyridine analog PAK-104P, which has been previously shown to reverse both ABCC1- and ABCB1-mediated drug resistance[37]. Sulindac sulfide inhibited [3H]-LTC4transport significantly at 10 μmol/L and in a dose dependent manner at concentrations similar to those which reversed resistance to chemotherapeutics (Fig. 5A). By contrast, the transport of LTC4into membrane vesicles from the parental line KB-3-1 was unaffected by sulindac sulfide. This data suggests that sulindac sulfide selectively inhibits ABCC1- mediated substrate transport.
Glutathione depletion in H69AR cells
Previous studies have shown that reduced glutathione (GSH) is either co-transported with or stimulates the transport of some substrates of ABCC1[38]. We therefore measured intracellular glutathione to determine if sulindac sulfide transport by ABCC1 as associated with glutathione depletion. As shown in Fig. 5B, sulindac sulfide significantly reduced glutathione levels in a concentration-dependent manner at levels that paralleled those that were effective for enhancing sensitivity to Dox. Furthermore, the combination of the glutathione synthetase inhibitor BSO with 10 μmol/L SS or 500 nmol/L Dox caused greater than additive depletion of glutathione (Fig. 5C). Combined treatment of sulindac sulfide and BSO resulted in highly potent suppression of the growth of ABCC1 expressing H69AR cells (Fig. 5D). This combined effect appeared to be specific for ABCC1 since the growth inhibitory activity of sulindac sulfide in HT-29 colon tumor cells, which express either low or no ABCC1 (or ABCB1), was not affected by combined treatment with BSO (Fig. 5E). These data suggest that sulindac sulfide reduces intracellular glutathione levels and that cells overexpressing ABCC1 may be moresusceptible to sulindac sulfide induced cytotoxicity by a mechanism involving glutathione depletion.
Sulindac sulfide increases sensitivity to Dox in a large panel of human cancer cells
To determine the prevalence of the Dox sensitizing effect, we performed similar dose-response studies with or without sulindac sulfide in a large panel of human tumor cell types. Totally, 47 human cell lines derived from a variety of cancer types were treated with 5 μmol/L SS or vehicle, followed by a concentration curve of doxorubicin for 72 hours. Dose-response curves were plotted for each, and IC50 values are presented (Table1). Changes in potency ranged from over 4-fold to less than 1-fold in the tumor cells of various histotypes. Of the 47 cell lines evaluated, 24 were more sensitive to Dox in the presence of a non-cytotoxic concentration (5 μmol/L) of sulindac sulfide that was statistically significance (e.g. no overlap in 95% confidence interval of the IC50value). The greatest sensitization occurred in OVCAR-5 ovarian cancer cells, with the IC50value decreasing from 0.542 μmol/L to 0.134 μmol/L, a 4-fold change, and two lung tumor cell lines (NCI-H322M and HOP-92) in which sensitivity was enhanced by 2.9-fold. Strikingly, all of the melanoma cell lines tested demonstrated significant sensitization to Dox following SS treatment.
Discussion
Here we show that the NSAIDs, indomethacin and sulindac sulfide can increase the sensitivity of ABCC1 overexpressing cells to chemotherapeutic drugs at concentrations that correspond to those achieved in the plasma with clinically relevant dosages[35]. Sulindac sulfide is the predominant species in the blood generated by enteric and hepatic reduction of the sulfoxide prodrug form of sulindac. In fact, sulindac sulfide was able to reverse ABCC1 mediated MDR and substrate transport at concentrations below its IC50for either COX-1 or COX-2[39]. Sulindac sulfide also significantly increased the intracellular accumulation and retention of Dox in vitro. Sulindac sulfide significantly decreased the accumulation of LTC4in inside-out membranes harboring ABCC1 and increased the fluorescence intensity of calcein-AM and Dox in ABCC1 overexpressing cells. Moreover, sulindac sulfide increased glutathione depletion in ABCC1 expressing cells in a dose dependent manner and further sensitized the cells to BSO and Dox treatment.
Previous studies have shown that certain NSAIDs are able to enhance the effects of some chemotherapeutic drugs in vitro[14]. These effects appear to be independent of COX-1 or COX-2 inhibition as the non- COX inhibitory sulfone metabolite also inhibits ABCC1 transport. We found that pretreatment of human lung cancer cells with sulindac sulfide enhanced their sensitivity to growth inhibition by Dox. The enhanced sensitivity to Dox was not observed in lung cancer cells expressing little or no ABCC1. Similar effects were apparent with a more potent COX inhibitor, indomethacin, although sulindac sulfide had a more pronounced effect on MDR reversal. The mechanism by which sulindac sulfide enhances the action of Dox is most likely independent of the suppression of COX, given that there is no correlation between the potency of COX inhibition and the sensitization to growth inhibition. In contrast, we observed a strong relationship between sulindac sulfide potency on acute effects of efflux compared with longer term effects on cell growth.
Sulindac sulfide displayed selectivity for ABCC1 as compared with ABCB1 and ABCG2 as shown by flow cytometry and LTC4uptake studies. This may have important implications for the potential clinical use of sulindac as a MDR reversal agent. While previous generations of ABC transport inhibitors have demonstrated toxicity, sulindac may be less toxic and accompanied with anticancer activity itself. The toxicity associated with previous MDR reversal agents has been attributed to the tissue distribution of ABCB1 and the effects of ABCB1 inhibitors on cytochrome P450 enzymes. Sulindac and its metabolites appear not to interfere with the cytochrome P450 enzymes or increase the toxicity among patients receiving epirubicin and sulindac in combination[35]. Although ABCC1 is found in tissues throughout the body, it is generally localized to the basolateral membrane. In comparison, both ABCB1 and ABCG2 are located in the apical membrane of cells such as colon epithelium and bile canalicular membranes[40]. Further complicating matters is the evidence that polymorphisms in ABCG2 can lead to unexpected anticancer drug interactions[41]. In contrast, transport mediated by ABCC1 seems relatively unaffected by such polymorphisms[42]. Thus, the selectivity of sulindac for ABCC1 indicates that it may have reduced toxicity when used in combination with chemotherapy.
ABCC1 is capable of transporting multiple substrate types, and several model systems are available to assay ABCC1 activity. In the present work, sulindac sulfide inhibited ABCC1-mediated transport of appropriate endogenous and xenobiotic substrates[43]. For example, the endogenous ABCC1 substrate, LTC4, is incorporated into isolated membrane vesicles isolated fromKB-CV60 cells and this activity was potently inhibited by sulindac sulfide. Consistent with previous reports in which calcein-AM efflux can be strongly correlated with ABCC1 expression and activity[44-45], we found that calcein-AM was excluded from ABCC1 expressing cells by both imaging and flow cytometry assays, and that this activity was also potently inhibited by sulindac sulfide. With the range of clinically important substrates for ABCC1 it is likely that compounds such as sulindac sulfide or derivatives have the potential to be useful in combination with different chemotherapeutic regimens to improve the clinical response to these drugs.
Although there is partial overlap of substrate specifi cities between ABCB1 and ABCC1, GSH conjugation or co-transport seems to be a requirement only for ABCC1 mediated transport. In contrast, GSH-conjugated organic anions are transported much less efficiently, if at all, by ABCB1. Consistent with a requirement for GSH to transport xenobiotics, growth inhibition of ABCC1 expressing cells by sulindac sulfide was increased nearly 7-fold in the presence of BSO, an inhibitor of the enzyme responsible for the rate-limiting step in GSH synthesis, gamma-glutamylcysteine synthetase. In contrast, sensitization to sulindac sulfide by BSO was not observed in cells expressing little or no ABCC1. Our data suggest that sulindac sulfide can sensitize ABCC1 expressing cells to further oxidative stress by decreasing intracellular glutathione levels. Although the interaction between ABCC1, anticancer drugs, and glutathione is not completely understood, it seems that most of the anticancer drugs to which ABCC1 confers resistance are not conjugated to GSH in vivo[46]. Instead, some of them are co-transported from cells with the reduced form of glutathione by ABCC1. Exploiting this distinction may lead to the development of selective inhibitors of MDR, especially for malignancies where ABCC1 seems to be the dominant cause of multidrug resistance, such as melanoma, glioma, and chronic lymphocytic leukemia[47-49].
In addition to the effects on ABCC1- mediated MDR described in the present studies, there are numerous reports which illustrate the cancer chemopreventive properties of non-aspirin NSAIDs, such as sulindac. Epidemiologic evidence supports the efficacy of sulindac for the prevention of colon and other cancers, particularly in the context of familial adenomatous polyposis[50-51]. Our lab has demonstrated that this effect is strongly correlated with inhibition of cGMP phosphodiesterase (PDE) activity[52-53]. Thus, the combination of sulindac with cytotoxic chemotherapeutics may provide a dual treatment benefit by inhibition of ABCC1-mediated efflux of cytotoxic compounds such as doxorubicin and direct inhibition of tumor cell growth by mechanisms independent of ABCC1, such as cGMP PDE inhibition.
Our data indicate that sulindac sulfide has a mechanism of action by which it not only inhibits ABCC1 mediated efflux of doxorubicin and other substrates leading to the intracellular accumulation of those substrates; it also depletes cells of GSH. The sensitivity of ABCC1 expressing cells to oxidative stress as seen in our experiments is in agreement with previously published data where either inhibition of GSH synthesis or increased GSH export preceded tumor cell apoptosis[54]. Based on the previous research of others and the data we have presented above, we believe the data is significant for the addition of sulindac to certain chemotherapeutic regimens. These studies also provide insight for the design of novel ABCC1 inhibitors by chemically modifying sulindac sulfide to improve potency and selectivity to inhibit ABCC1-mediated efflux for preventing drug resistance and tumor recurrence or secondary tumor formation follow ing chemotherapy.
Acknowledgments
We thank Irena Ivnitski-Steele for flow cytometry analysis. ABCB1 and ABCC1 expressing cells were kindly provided by Richard S. Larson, MD, PhD, University of New Mexico. We thank Dr. D. Ross from the Department of Medicine, University of Maryland Greenebaum Cancer Center, Baltimore, MD) for providing ovarian Ig-MXP3 (ABCG2) and its parental Igrov1-sensitive cells.
Financial Support: NIH grants CA131378 (GAP), CA148817 (GAP), CA155638 (GAP), U54HG-003917 (GAP), U54MH084690 (LAS), and U54-MH074425 (LAS).
References
[1] Thomas, H, Coley, HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein[J]. Cancer Control, 2003,10:159-165.
[2] Deeley, RG, Westlake, C, Cole, SP. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins[J]. Physiol Rev, 2006,86:849-899.
[3] Juliano, RL, Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants[J]. Biochim Biophys Acta, 1976,455:152-162.
[4] Cole, SP, Bhardwaj, G, Gerlach, JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line[J]. Science, 1992,258:1650-1654.
[5] Endicott, J, ALing, V. The biochemistry of P-glycoproteinmediated multidrug resistance[J]. Annu Rev Biochem, 1989,58:137-171.
[6] Borst, P, Evers, R, Kool, M, et al. A family of drug transporters: the multidrug resistance-associated proteins[J]. J Natl Cancer Inst, 2000,92:1295-1302.
[7] Rosenbaum, C, Rohrs, S, Muller, O, et al. Modulation of MRP-1-mediated multidrug resistance by indomethacin analogues[J]. J Med Chem, 2005,48:1179-1187.
[8] Doyle, LA, Yang, W, Abruzzo, LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells[J]. Proc Natl Acad Sci U S A, 1998,95:15665-15670.
[9] Shukla, S, Wu, CP, Ambudkar, SV. Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges[J]. Expert Opin Drug Metab Toxicol, 2008,4:205-223.
[10] Choi, YH, Yu, AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development, Netherlands: 793-807.
[11] Leveque, D, Jehl, F. P-glycoprotein and pharmacokinetics[J]. Anticancer Res, 1995,15:331-336.
[12] Lin, JH, Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: clinical implications[J]. Clin Pharmacokinet, 2003,42:59-98.
[13] Evers, R, Zaman, GJ, van Deemter, L, et al. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells[J]. J Clin Invest, 1996,97:1211-1218.
[14] Duffy, CP, Elliott, CJ, O'Connor, RA, et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti- inflammatory drugs (NSAIDs)[J]. Eur J Cancer, 1998,34:1250-1259.
[15] Benyahia, B, Huguet, S, Decleves, X, et al. Multidrug resistance-associated protein MRP1 expression in human gliomas: chemosensitization to vincristine and etoposide by indomethacin in human glioma cell lines overexpressing MRP1[J]. J Neurooncol, 2004,66:65-70.
[16] O'Connor, R, Heenan, M, Connolly, L, et al. Increased anti-tumour efficacy of doxorubicin when combined with sulindac in a xenograft model of an MRP- 1-positive human lung cancer[J]. Anticancer Res, 2004,24:457-464.
[17] Trifan, OC, Durham, WF, Salazar, VS, et al. Cyclooxygenase-2 inhibition with celecoxib enhances antitumor efficacy and reduces diarrhea side effect of CPT-11[J]. Cancer Res, 2002,62:5778- 5784.
[18] O'Connor, R, O'Leary, M, Ballot, J, et al. A phase I clinical and pharmacokinetic study of the multi-drug resistance protein-1 (MRP-1) inhibitor sulindac, in combination with epirubicin in patients with advanced cancer[J]. Cancer Chemother Pharmacol, 2007,59:79-87.
[19] Piazza, GA, Rahm, AK, Finn, TS, et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction[J]. Cancer Res, 1997,57:2452- 2459.
[20] Piazza, GA, Rahm, AL, Krutzsch, M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis[J]. Cancer Res, 1995,55:3110-3116.
[21] Estes, DA, Lovato, DM, Khawaja, HM, et al. Genetic alterations determine chemotherapy resistance in childhood T- ALL: modelling in stage-specific cell lines and correlation with diagnostic patient samples[J]. Br J Haematol, 2007,139:20-30.
[22] Winter, SS, Jiang, Z, Khawaja, HM, et al. Identification of genomic classifiers that distinguish induction failure in T-lineage acute lymphoblastic leukemia: a report from the Children's Oncology Group[J]. Blood, 2007,110:1429-1438.
[23] Monga, M, Sausville, EA. Developmental therapeutics program at the NCI: molecular target and drug discovery process[J]. Leukemia, 2002,16:520-526.
[24] Collins, JM. The NCI Developmental Therapeutics Program[J]. Clin Adv Hematol Oncol, 2006,4:271-273.
[25] Aoki, S, Chen, ZS, Higasiyama, K, et al. Reversing effect of agosterol A, a spongean sterol acetate, on multidrug resistance in human carcinoma cells[J]. Jpn J Cancer Res, 2001,92:886-895.
[26] Ivnitski-Steele, I, Larson, RS, Lovato, DM, et al. High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters[J]. Assay Drug Dev Technol, 2008,6:263-276.
[27] Winter, SS, Lovato, DM, Khawaja, HM, et al. Highthroughput screening for daunorubicin-mediated drug resistance identifies mometasone furoate as a novel ABCB1-reversal agent[J]. J Biomol Screen, 2008,13: 185-193.
[28] Mirski, SE, Gerlach, JH, Cole, SP. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin[J]. Cancer Res, 1987,47:2594-2598.
[29] Harker, W, GSikic, BI. Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA[J]. Cancer Res, 1985,45:4091-4096.
[30] Cole, SP, Sparks, KE, Fraser, K, et al. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells[J]. Cancer Res, 1994,54:5902-5910.
[31] Harker, WG, Bauer, D, Etiz, BB, et al. Verapamilmediated sensitization of doxorubicin-selected pleiotropic resistance in human sarcoma cells: selectivity for drugs which produce DNA scission[J]. Cancer Res, 1986, 46:2369-2373.
[32] Gosland, MP, Lum, BL, Sikic, BI. Reversal by cefoperazone of resistance to etoposide, doxorubicin, and vinblastine in multidrug resistant human sarcoma cells[J]. Cancer Res, 1989,49:6901-6905.
[33] Creasey, WA, McIntosh, LS, Brescia, T, et al. Clinical effects and pharmacokinetics of different dosage schedules of adriamycin[J]. Cancer Res, 1976,36:216- 221.
[34] Chang, JK, Wang, GJ, Tsai, ST, et al. Nonsteroidal antiinflammatory drug effects on osteoblastic cell cycle, cytotoxicity, and cell death[J]. Connect Tissue Res, 2005, 46:200-210.
[35] Davies, NM, Watson, MS. Clinical pharmacokinetics of sulindac. A dynamic old drug[J]. Clin Pharmacokinet, 1997,32:437-459.
[36] Leier, I, Jedlitschky, G, Buchholz, U, et al. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates[J]. J Biol Chem, 1994,269:27807-27810.
[37] Vanhoefer, U, Cao, S, Minderman, H, et al. PAK-104P, a pyridine analogue, reverses paclitaxel and doxorubicin resistance in cell lines and nude mice bearing xenografts that overexpress the multidrug resistance protein[J]. Clin Cancer Res, 1996,2:369-377.
[38] Jedlitschky, G, Leier, I, Buchholz, U, et al. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump[J]. Cancer Res, 1996,56:988-994.
[39] Whitt, JD, Li, N, Tinsley, HN, et al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and betacatenin transcriptional activity. Cancer Prev Res (Phila) 5:822-833.
[40] Doyle, LA, Ross, DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2)[J]. Oncogene, 2003,22:7340-7358.
[41] Noguchi, K, Katayama, K, Mitsuhashi, J, et al. Functions of the breast cancer resistance protein (BCRP/ABCG2) in chemotherapy[J]. Adv Drug Deliv Rev, 2009,61:26-33.
[42] Cascorbi, I. Role of pharmacogenetics of ATP-binding cassette transporters in the pharmacokinetics of drugs[J]. Pharmacol Ther, 2006,112:457-473.
[43] Strouse, JJ, Ivnitski-Steele, I, Waller, A, et al. Fluorescent substrates for flow cytometric evaluation of efflux inhibition in ABCB1, ABCC1, and ABCG2 transporters[J]. Analytical biochemistry 437:77-87.
[44] Legrand, O, Simonin, G, Perrot, JY, et al. Pgp and MRP activities using calcein-AM are prognostic factors in adult acute myeloid leukemia patients[J]. Blood, 1998,91:4480-4488.
[45] Olson, DP, Taylor, BJ, Ivy, SP. Detection of MRP functional activity: calcein AM but not BCECF AM as a Multidrug Resistance-related Protein (MRP1) substrate[J]. Cytometry, 2001,46:105-113.
[46] Leslie, EM, Deeley, RG, Cole, SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters[J]. Toxicology, 2001, 167:3-23.
[47] Walsh, N, Kennedy, S, Larkin, AM, et al. Membrane transport proteins in human melanoma: associations with tumour aggressiveness and metastasis[J]. Br J Cancer 102:1157- 1162.
[48] Spiegl-Kreinecker, S, Buchroithner, J, Elbling, L, et al. Expression and functional activity of the ABC-transporter proteins P-glycoprotein and multidrug- resistance protein 1 in human brain tumor cells and astrocytes[J]. J Neurooncol, 2002,57:27- 36.
[49] Fazlina, N, Maha, A, Zarina, AL, et al. Assessment of Pgp and MRP1 activities using MultiDrugQuant Assay Kit: a preliminary study of correlation between protein expressions and its functional activities in newly diagnosed acute leukaemia patients[J]. Malays J Pathol, 2008,30: 87-93.
[50] Brasky, TM, Liu, J, White, E, et al. Non-steroidal antiinflammatory drugs and cancer risk in women: results from the Women's Health Initiative. International journal of cancer[J]. Journal international du cancer 135:1869-1883.
[51] Kim, B, Giardiello, FM. Chemoprevention in familial adenomatous polyposis, Netherlands: 2011 Elsevier Ltd, 2011:607-622.
[52] Tinsley, HN, Gary, BD, Thaiparambil, J, et al. Colon tumor cell growth-inhibitory activity of sulindac sulfide and other nonsteroidal anti-inflammatory drugs is associated with phosphodiesterase 5 inhibition[J]. Cancer Prev Res (Phila) 3:1303-1313.
[53] Gurpinar, E, Grizzle, WEPiazza, GA. NSAIDs inhibit tumorigenesis, but how?[J] Clin Cancer Res 20:1104-1113.
[54] Cole, SP, Downes, HF, Mirski, SE, et al. Alterations in glutathione and glutathione-related enzymes in a multidrug-resistant small cell lung cancer cell line[J]. Mol Pharmacol, 1990,37:192-197.
✉ Gary A. Piazza, Ph.D., Chief ofDrugDiscovery, Professor of Oncologic Sciences and Pharmacology, Abraham A. Mitchell Distinguished Investigator, USA Mitchell Cancer Institute, University of SouthAlabama 1660 Springhill Avenue, Suite 3029, Mobile AL 36604, 251-445-8412, E-mail: gpiazza@health.southalabama.edu.
03 August 2015, Revised 25 August 2015, Accepted 26 October 2015, Epub 20 November 2015
R969.1, Document code: A
The authors reported no conflicts of interests.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Organ donation in China: the major progress and the continuing problem
- Cerebral ischemia during surgery: an overview
- Apolipoprotein A-V gene therapy for disease prevention / treatment: a critical analysis
- HDL signaling and protection against coronary artery atherosclerosis in mice
- Prevalence and risk factors of HIV and syphilis, and knowledge and risk behaviors related to HIV/AIDS among men who have sex with men in Chongqing, China
- Effects of closing and reopening live poultry markets on the epidemic of human infection with avian influenza A virus