HDL signaling and protection against coronary artery atherosclerosis in mice
2016-12-13BernardoTrigattiMarkFuller
Bernardo L Trigatti, Mark Fuller
Department of Biochemistry and Biomedical Sciences, McMaster University and Thrombosis and Atherosclerosis Research Institute, McMaster University and Hamilton Health Sciences., Hamilton, Ontario L8L 2X2, Canada.
HDL signaling and protection against coronary artery atherosclerosis in mice
Bernardo L Trigatti✉, Mark Fuller
Department of Biochemistry and Biomedical Sciences, McMaster University and Thrombosis and Atherosclerosis Research Institute, McMaster University and Hamilton Health Sciences., Hamilton, Ontario L8L 2X2, Canada.
Atherosclerosis is a leading underlying factor in cardiovascular disease and stroke, important causes of morbidity and mortality across the globe. Abundant epidemiological studies demonstrate that high levels of high density lipoprotein (HDL) are associated with reduced risk of atherosclerosis and preclinical, animal model studies demonstrate that this association is causative. Understanding the molecular mechanisms underlying the protective effects of HDL will allow more strategic approaches to development of HDL based therapeutics. Recent evidence suggests that an important aspect of the ability of HDL to protect against atherosclerosis is its ability to trigger signaling responses in a variety of target cells including endothelial cells and macrophages in the vessel wall. These signaling responses require the HDL receptor, scavenger receptor class B type 1 (SR-B1), an adaptor protein (PDZK1) that binds to the cytosolic C terminus of SR-B1, Akt1 activation and (at least in endothelial cells) activation of endothelial NO synthase (eNOS). Mouse models of atherosclerosis, exemplified by apolipoprotein E or low density lipoprotein receptor gene inactivated mice (apoE or LDLR KO) develop atherosclerosis in their aortas but appear generally resistant to coronary artery atherosclerosis. On the other hand, inactivation of each of the components of HDL signaling (above) in either apoE or LDLR KO mice renders them susceptible to extensive coronary artery atherosclerosis suggesting that HDL signaling may play an important role in protection against coronary artery disease.
coronary artery disease, high density lipoprotein, myocardial infarction, scavenger receptor class B type 1
Introduction
Abundant epidemiological studies in populations and preclinical studies in animal models demonstrate that high density lipoprotein (HDL) protects against the development of atherosclerosis. The scavenger receptor class B type 1 (SR-B1) is a multiligand receptor that has high affinity for HDL and can mediate the bi-directional exchange of lipids between bound HDL and cells[1]. SR-B1 expressed in hepatocytes mediates cellular uptake of cholesterol from HDL, driving a process called reverse cholesterol transport: the net movement of cholesterol by HDL from peripheral tissues including macrophages in the artery wall, to the liver for disposal via the bile or for recycling back into the bloodstream complexed with new lipoproteins[1]. In addition to this well accepted role for SR-B1 in cholesterol transport, more recent studies have pointed toa role in HDL-dependent signaling in cells including endothelial cells, macrophages, platelets and other cell types[2-12]. We have recently demonstrated that HDL acts as a chemotactic factor for macrophages in vitro, and that this involves HDL dependent signaling via SR-B1, PDZK1 and sphingosine 1 phosphate (S1P) receptor 1 (S1PR1), leading to phosphorylation of Akt[2]. Inactivation of the expression of SR-B1, PDZK1 or Akt1 or antagonism of S1PR1 impairs the ability of macrophages to undergo chemotaxis towards HDL[2]. Similarly, others have demonstrated that HDL signals via SR-B1 in platelets and in hematopoietic stem/progenitor cells (HSPC) leading to PI3K/Akt activation, which dampens platelet activation and HSPC proliferation[4-6]. HDL signaling has been most well studied in endothelial cells, in which HDL signaling via SR-B1 leads to phosphorylation of PI3K/Akt, and ofendothelial NO synthase (eNOS) leading to its activation (Fig. 1). HDL signaling in endothelial cells also results in cell migration and proliferation, and suppresses activation of expression of the inflammatory adhesion molecules vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1)[3,7-8,10,12].
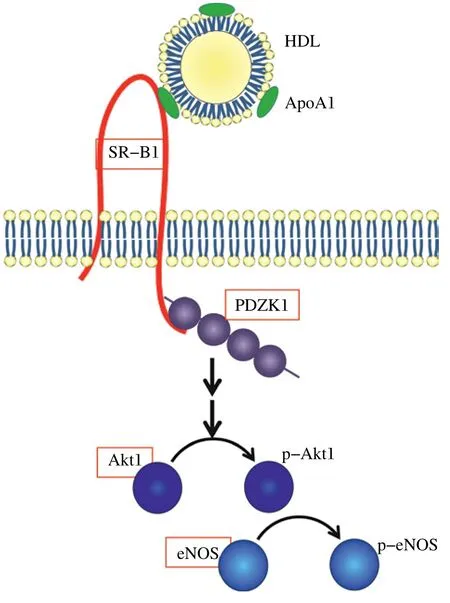
Fig. 1 HDL signaling via SR-B1 and role in coronary artery atherosclerosis. HDL signaling via SR-B1 involves PDZK1 (shown binding to the C-terminal tail of SR-B1 via one of the two possible PDZ domains known to bind there), activation of Akt1 and eNOS. Germline whole body knockout of SR-B1, PDZK1, Akt1 and eNOS (red boxes) on atherogenic ApoE or LDLR KO backgrounds have all been shown to trigger either spontaneous (SR-B1/apoE) or diet induced coronary artery atherosclerosis and myocardial infarction (PDZK1, Akt1 or eNOS on apoE KO; SR-BI on apoE-hypo or LDLR KO backgrounds)[19,21,23,31-33]. More detailed descriptions of HDL signaling pathways mediated by SR-B1 can be found in references[8-9].
VCAM-1 and ICAM-1 play key roles in adhesion of monocytes to activated endothelial cells of arteries at sites prone to atherosclerosis development[13-14]. Endothelial cells can become activated to express high levels of VCAM-1 and ICAM-1 through signaling induced by inflammatory cytokines such as tumor necrosis factor (TNF) α, or by non-laminar, turbulent blood flow; the latter being a key determinant of the location of atherosclerosis development within the arterial tree[7,13-14]. Exposure of monocytes to HDL has been reported to suppress the VCAM-1 binding activity of CD49d and the ICAM-1 binding activity of CD11b, thereby reducing monocyte recruitment by activated endothelial cells in vitro[15-16]. These findings suggest that HDL induces atheroprotective cellular responses locally in the blood vessel/at the sites of lesions that are dependent on SR-B1 but may be independent of bulk cholesterol transport.
SR-B1 and atherosclerosis
To investigate the role of SR-B1 on atherosclerosis, SR-B1 deficient mice were generated and mated to apolipoprotein (apo) E deficient mice to generate SR-B1/apoE double knockout (dKO) mice. These mice develop increased aortic sinus atherosclerosis compared with control apoE KO mice when fed a normal chow diet[17]. Surprisingly, these mice also develop occlusive atherosclerosis in a substantial proportion (approximately 30%) of coronary arteries, accompanied by extensive myocardial fibrosis, electrical conductance abnormalities including ST segment deviations, cardiac dysfunction characterized by reduced left ventricular ejection fraction and early death by approximately 8 weeks of age when fed a normal chow diet[18-20]. Krieger and co-workers also generated a related strain of mice which lacked SR-B1 on a hypomorphic (hypo) apoE background[21]. These SR-B1-KO/apoE-hypo mice appear normal when fed a normal chow diet, low in fat and cholesterol; however feeding the mice diets containing high fat, high cholesterol and sodium cholate induces the development of occlusive coronary artery atherosclerosis and greater aortic sinus atherosclerosis as compared to similarly fed SR-B1 expressing apoE-hypo mice[21]. The induction of occlusive atherosclerosis in coronary arteries is rapid, requiring only 3-4 weeks, and, as in SR-B1/apoE dKO mice, is accompaniedby extensive myocardial fibrosis, cardiomegaly, ST segment deviations in electrocardiograms, and left ventricular dysfunction[21]. The timing of disease development in the SR-B1-KO/apoE-hypo mice, as measured by survival and cardiomegaly, is affected by the composition of the diet, such that increased dietary cholesterol and the presence of sodium cholate, a bile salt derivative which results in increased levels of plasma cholesterol, result in more rapid onset of cardiomegaly and reduced survival[22]. Husbandry conditions can also affect the survival of the atherogenic, high fat/high cholesterol diet-fed SR-B1-KO/apoE-hypo mice, such that survival is prolonged when mice are group-housed vs. housed singly[22]. Surprisingly, mice housed in groups were reported to exhibit lower levels of plasma cholesterol than those housed singly, although the reasons for this appear unclear[22]. However, it was suggested that this might contribute to the prolonged survival of the group-housed atherogenic diet fed SR-B1/apoE-hypo mice[22]. The effects of the different diets on the level of atherosclerosis in coronary arteries or the aortic sinus, however was not described.
Coronary artery atherosclerosis in SR-B1/ LDLR double KO mice
We have recently reported that SR-B1/LDLR dKO mice also exhibit reduced survival when fed atherogenic diets, and, similar to effects observed in SR-B1-KO/apoE-hypo mice, the average lifespan of atherogenic diet-fed SR-B1/LDLR dKO mice was reduced by increased cholesterol content, and presence of sodium cholate in the diet[23]. When female SR-B1/LDLR dKO mice were fed the Paigen atherogenic diet (15% fat, 1.25% cholesterol, 0.5% sodium cholate) beginning at 12 weeks of age, they exhibited a 50% survival of only 3.5 weeks from the onset of feeding[23], similar to the median survival reported for the SR-B1-KO/apoE-hypo mice fed the same diet[21,22]. Removal of sodium cholate prolonged the 50% survival to 9.4 weeks of diet feeding, while mice fed a normal chow diet supplemented with 2% cholesterol exhibited a 50% survival of 11.4 weeks[23]. In contrast SR-B1/ LDLR dKO mice fed a Western type atherogenic diet (21% fat and 0.15% cholesterol) from 12 weeks of age, or a normal chow diet (6.2% fat, and no added cholesterol) from weaning did not exhibit any reduced survival up to 22 weeks of age[23]. For each diet/feeding time, female SR-B1/LDLR dKO mice exhibited statistically significantly increased (3.5-10 fold) atherosclerosis in their aortic sinuses when compared with similarly aged and fed female LDLR single KO mice[23-24]. SR-B1/LDLR dKO mice fed each of the atherogenic diets, but not mice fed the normal chow diet, developed significant coronary artery atherosclerosis (Fig. 2), with approximately 20% (Western diet) to 30% (Paigen diet) of coronary arteries on average, being completely occluded by atherosclerotic plaques[23]. In contrast, virtually none of the coronary arteries examined in any of the atherogenic diet fed LDLR single KO control mice contained atherosclerotic plaques (although some fatty streaks were detected in Paigen diet fed LDLR KO mice)[23]. Furthermore, mice fed the cholesterol-supplemented diet, the Paigen diet and the Paigen diet lacking sodium cholate all exhibited substantial levels of cardiac fibrosis and cardiomegaly compared to age, gender and diet matched control LDLR single KO's. Surprisingly, the SR-B1/LDLR dKO mice fed the Western type diet for 12 weeks did not develop substantial cardiac fibrosis, although they did develop cardiomegaly to a similar degree as mice fed the other atherogenic diets[23]. Interestingly, we detected platelet accumulation in coronary artery atherosclerotic plaques in the atherogenic diet fed SR-B1/LDLR dKO mice, and the abundance of platelet-positive atherosclerotic coronary arteries was highest in mice fed the Paigen atherosclerotic diet (Fig. 2B), which resulted in the most rapid reduction in survival[23]. Whether the accumulation of platelets in atherosclerotic coronary arteries in the Paigen diet fed SR-B1/LDLR dKO mice is the result of thrombosis secondary to plaque rupture remains to be determined. Together these data demonstrate the diet dependence of coronary artery atherosclerosis and cardiac fibrosis in the SR-B1/LDLR dKO mice.
Levels of coronary artery atherosclerosis do not correlate with levels of aortic sinus atherosclerosis
Interestingly, we observed that the levels of coronary artery atherosclerosis did not correlate with the levels of aortic sinus atherosclerosis in the SR-B1/ LDLR dKO and LDLR single KO mice fed the five different diets. In particular, we observed similar levels of aortic sinus atherosclerosis in 22 week old normal chow diet fed SR-B1/LDLR dKO mice and 13.5 week old SR-B1/LDLR dKO mice that had been fed the Paigen atherogenic diet for 3.5 weeks[23]. The levels of aortic sinus atherosclerosis in these mice were also similar to those in LDLR single KO mice fed the 2% cholesterol supplemented diet for 12 weeks or fed the Paigen atherogenic diet lacking sodium cholate for 10 weeks. Despite similar levels of aortic sinus atherosclerosis, the only group of these four to develop severe coronary artery atherosclerosis werethe SR-B1/LDLR double KO mice fed the Paigen diet for 3.5 weeks-no coronary artery atherosclerosis was detected in LDLR single KO mice fed either of the high cholesterol diets and very little was detected in the SR-B1/LDLR dKO mice fed the normal chow diet for the times indicated[23]. This observation clearly demonstrated that coronary artery atherosclerosis was not merely a manifestation of increased overall atherosclerosis burden, but rather suggests different factors determined the development of atherosclerosis in the aortic sinus and coronary arteries of these mice.
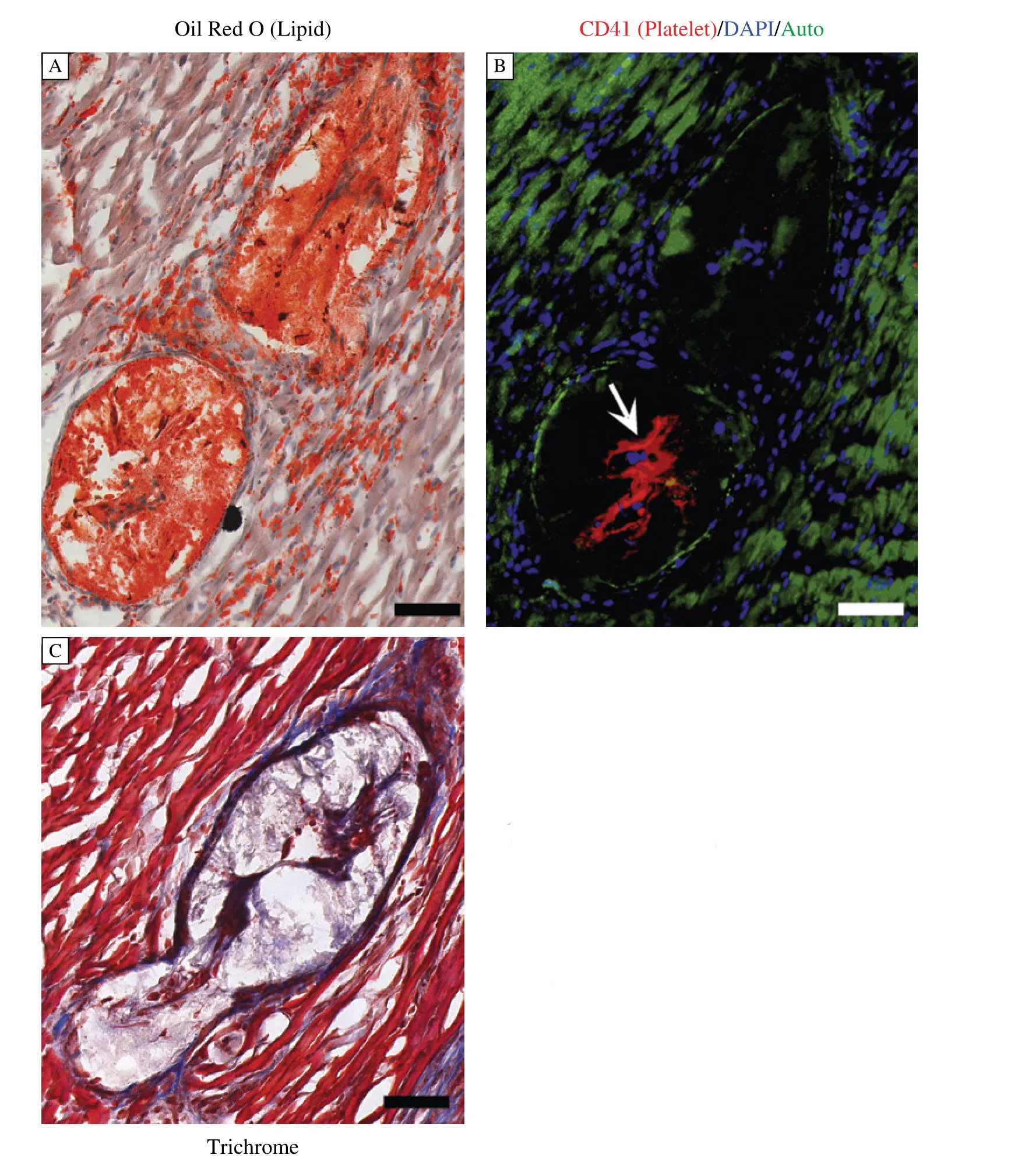
Fig. 2 Occlusive coronary artery atherosclerosis in an atherogenic diet fed SR-B1/LDLR double KO mouse: In an experiment described in(23), an SR-B1/ LDLR double KO mouse was fed the Paigen atherogenic diet for 3.5 weeks. Shown is a series of cross sections through the same coronary artery (passing through the septum of the heart), just below (A) and (B) and at (C) a bifurcation. (A) Oil red O (lipid-red)/hematoxylin (nuclei-blue) stained section. (B) Immunofluorescence staining for CD41 (platelet-red); counterstained with DAPI (nuclei-blue). Green: autofluorescence. Arrow points to red CD41 staining. (C) Trichrome staining. Fibrous tissue appears blue. Scale bar = 30 μm.
We observed that the majority of the non-atherosclerotic coronary arteries (75%-100%) analyzed in the atherogenic diet fed SR-B1/LDLR dKO mice exhibited high levels of VCAM-1 and ICAM-1 staining, whereas substantially fewer coronary arteries from similarly fed LDLR single KO mice were positive for VCAM-1 and ICAM-1[23]. This suggests that one consequence of inactivating SR-B1 expression is increased activation of coronary artery endothelial cells to express these adhesion molecules, which are known to mediate monocyte adhesion and recruitment into the vessel wall, and influence the sites of atherosclerosis development[13-14]. These results are consistent with the finding that SR-B1 in endothelial cells mediates HDL dependent suppression of VCAM-1 and ICAM-1 induction by TNF-α[7]. Whether or not the increased VCAM-1 and ICAM-1 in coronary arteries of the SR-B1/LDLR dKO mice is thedirect effect of a lack of SR-B1 expression in endothelial cells, or is an indirect effect of, for example, increased levels of IL-6 and/or TNF-α in circulation, or altered properties/levels of circulating lipoproteins[23], remains unclear. Although total plasma cholesterol levels were lower in the atherogenic diet fed SR-B1/LDLR dKO mice (corresponding to reduced hepatic lipoprotein production), the lipoproteins present in plasma of the dKO mice were characterized by abnormally high unesterified cholesterol (reaching 50%-80% of total cholesterol) in the SR-B1/LDLR dKO mice as compared to the lower levels of unesterified cholesterol (25%-27% of total cholesterol) in LDLR single KO mice[23], consistent with what has been previously described for SR-B1/apoE dKO and SR-B1 KO/apoE-hypo mice[18,20-22,25]. The effects of altered lipoprotein composition and structure in SR-B1/LDLR dKO mice on endothelial cell activation, however, has not been examined directly.
Others have reported that HDL treatment of monocytes can suppress monocyte binding to VCAM-1 and ICAM-1 and monocyte adhesion to endothelial cells[15-16]. Consistent with this, we have demonstrated that monocytes from SR-B1 KO/apoE-hypo mice exhibit greater binding to VCAM-1 and ICAM-1 and that restoration of SR-B1 expression in bone marrow-derived cells in SR-B1 KO/apoE-hypo mice resulted in reduced monocyte recruitment into atherosclerotic plaques and reduced aortic sinus and coronary artery atherosclerosis in mice fed the Paigen atherogenic diet[26]. This also led to reduced diet-dependent development of myocardial fibrosis and cardiomegaly[26]. In contrast, BM-specific restoration of SR-B1 expression did not restore altered lipoprotein cholesterol levels, raising the possibility that the protective effects reflected the direct activity of SR-B1 in monocytes or other BM derived cells.
In vivo evidence for signaling via SR-BI contributing to protection against coronary artery atherosclerosis?
HDL triggers the activation (phosphorylation) of Akt and Akt dependent phosphorylation and activation of eNOS in endothelial cells both in culture and in vivo[8,27]. This signaling process is dependent on SR-B1 and the multi-subunit adaptor protein, PDZK1, that binds to the carboxy terminal cytosolic domain of SR-B1[3,10,27](Fig. 1). PDZK1 is required for the stability of SR-B1 protein in the liver and inactivation of PDZK1 in mice results in dramatically reduced levels of SR-B1 protein in liver hepatocytes, but does not alter SR-B1 protein levels in endothelial cells or macrophages[2-3,12,28-30]. Instead, in endothelial cells and macrophages, PDZK1 appears to be necessary for SR-B1 mediated HDL signaling[2-3,12]. Curiously, global knockout of each of PDZK1, Akt1 or eNOS on the apoE KO background all lead to diet-induced coronary artery atherosclerosis and myocardial fibrosis, resembling the phenotype of SR-B1/apoE dKO mice and atherogenic diet fed SR-B1/apoE-hypo and SR-B1/LDLR dKO mice (although the degree of coronary artery atherosclerosis, myocardial fibrosis or effects on survival do not appear to be as severe as those observed in mice lacking SR-B1)[31-33](Fig. 1). Thus, Krieger, Kocher and colleagues demonstrated that feeding PDZK1/apoE dKO mice a high fat, low cholesterol Western diet (21% fat, 0.15% cholesterol) increased aortic sinus but not coronary artery atherosclerosis[29], but feeding the mice the Paigen diet for 3 months induced substantial coronary artery atherosclerosis (with up to 40% of coronary arteries examined containing plaques that occluded more than half of the lumen) and myocardial fibrosis[33]. These mice exhibited increased lipoprotein total cholesterol compared with similarly fed apoE KO mice but did not exhibit the increased lipoprotein unesterified cholesterol content characteristic of SR-B1 KO mice, possibly because low residual levels of SR-B1 protein in liver may have been sufficient to clear unesterified cholesterol normally[33]. HDL treatment of endothelial cells and macrophages in culture induces Akt phosphorylation and pharmacological inhibition of Akt phosphorylation or knockout of Akt1 prevents HDL signaling in these cells[2,34]. Akt1/ApoE dKO mice have been reported to develop increased aortic atherosclerosis compared to similarly fed apoE KO mice, and, reminiscent of SR-B1-deficient and PDZK1-deficient apoE KO mice, also develop high fat diet induced occlusive coronary artery atherosclerosis[31]. Furthermore, high fat diet feeding (16 weeks) was also reported to induce coronary artery atherosclerosis (in addition to increased aortic atherosclerosis), cardiac fibrosis and reduced cardiac function in eNOS/ apoE double KO mice, but not in apoE KO mice fed the same diet for up to 20 weeks[32]. It is important to point out that the phenotypes reported for SR-B1/ apoE, PDZK1/apoE, Akt1/apoE and eNOS/apoE double KO mice do not match exactly: SR-B1/apoE dKO mice develop occlusive coronary artery atherosclerosis spontaneously and rapidly (within 2-5 weeks) on normal chow diet[18-19]while PDZK1/apoE, Akt1/ apoE and eNOS/apoE dKO mice all require feeding with atherogenic diets (with different concentrations of fat, cholesterol and cholate)[29,31-33]. Furthermore, a subset of high fat diet fed eNOS/apoE dKO mice also develop abdominal aortic aneurysms, which has not been reported in SR-B1-deficient (SR-B1/apoE dKO, or in SR-B1KO/apoE-hypo or SR-B1/LDLRdKO mice fed atherogenic diet diets), or PDZK1/ apoE or Akt1/apoE dKO mice fed atherogenic diets[2,19,21,23,26,29,31-33]. To be sure, coronary artery atherosclerosis in mice does not solely arise from inactivation of components of the HDL/SR-B1/PDZK1/ Akt1/eNOS signaling pathway. Substantial coronary artery atherosclerosis accompanied by myocardial infarction has also been observed in atherogenic diet fed apoE KO mice with macrophage overexpression of urokinase type plasminogen activator[35-36]and in long term atherogenic diet fed LDL receptor/apoE double knockout mice[37]. Nevertheless, the finding that SR-B1, PDZK1, Akt1 and eNOS are linked by HDL signaling and the observation that atherogenic strains that lack these factors develop either spontaneous or diet-induced coronary artery atherosclerosis, suggests the possibility that impaired HDL signaling may contribute to the development of coronary artery atherosclerosis in these mice. It is tempting to speculate that the observed spontaneous or diet-induced coronary artery atherosclerosis in apoE and/or LDLR KO mice also lacking SR-B1, PDZK1, Akt1 or eNOS may reflect impaired HDL signaling in endothelial cells, resulting in increased endothelial cell activation in normally resistant coronary arteries. This is consistent with our observation of only very low levels of VCAM-1 and ICAM-1 protein in coronary arteries of high fat diet fed LDLR KO mice, and substantially increased levels in coronary arteries of similarly fed SR-B1/LDLR dKO mice[23]. Whether this is in fact due to impaired HDL signaling in endothelial cells (as predicted from in vitro studies cited earlier) or due other factors resulting from impaired global SR-B1 expression (e.g. altered lipoprotein structure, composition or levels; altered systemic inflammation; altered stress response etc) is currently unclear. Resolution of this question awaits the analysis of mice with tissue selective KO or re-expression of SR-BI.
Conclusions
HDL signaling via SR-B1, PDZK1 and Akt1 appears to play an important role in homeostatic and/ or atheroprotective responses in a variety of cell types including endothelial cells, macrophages and monocytes, HSPC's, smooth muscle cells and others[8-9]. In endothelial cells, this signaling pathway leads to activation of eNOS and a variety of atheroprotective outcomes including proliferation, recovery after injury, increased barrier function and suppression of endothelial cell VCAM-1 and ICAM-1 expression[3,7-10,12,27]. Global inactivation of SR-B1, PDZK1, Akt1 and eNOS in mice predisposed to atherosclerosis due to inactivation of apoE and/or LDLR, all lead to spontaneous or diet-induced coronary artery atherosclerosis and myocardial infarction[19,21,23,31-33]. These data suggest that HDL signaling via an SR-B1/PDZK1/Akt1/ eNOS axis plays an important role, at least in mice, in the normal resistance of coronary arteries to development of atherosclerosis. HDL signaling may explain, at least in part, the protective role that HDL exerts on human coronary artery disease. A better understanding of HDL signaling pathways, including the key cell types responsible for HDL signaling-mediated protection against coronary artery atherosclerosis in mice may provide important insights into strategies for therapeutic interventions in human coronary artery disease.
Acknowledgements
Research in the Trigatti lab is supported by funds from the Canadian Institutes of Health Research (MOP74765), and the Heart and Stroke Foundation of Canada (G-13-0002833 and G-15-0009016).
References
[1] Krieger M. Lipoprotein receptor SR-BI[J]. Annu Rev Biochem, 1999,68: 523-558.
[2] Al-Jarallah A, Chen X, Gonzalez L, et al. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists[J]. PLoS One,2014,9:e106487.
[3] Assanasen C, Mineo C, Seetharam D, et al. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling[J]. J Clin Invest, 2005;115(4):969-977.
[4] Brill A, Yesilaltay A, De Meyer SF, et al. Extrahepatic high-density lipoprotein receptor SR-BI and apoA-I protect against deep vein thrombosis in mice[J]. Arterioscler Thromb Vasc Biol, 2012,32: 1841-1847.
[5] Brodde MF, Korporaal SJ, Herminghaus G, et al. Native high-density lipoproteins inhibit platelet activation via scavenger receptor BI: role of negatively charged phospholipids[J]. Atherosclerosis, 2011,215:374-382.
[6] Gao M, Zhao D, Schouteden S, et al. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression[J]. Arterioscler Thromb Vasc Biol, 2014,34:1900-1909.
[7] Kimura T, Tomura H, Mogi C, et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells[J]. J Biol Chem, 2006,281:37457-37467.
[8] Mineo C, Shaul PW. Regulation of signal transduction by HDL[J]. J Lipid Res,2013,54: 2315-2324.
[9] Nofer JR. Signal transduction by HDL: agonists, receptors, and signaling cascades[J]. Handb Exp Pharmacol, 2015,224:229-256.
[10] Seetharam D, Mineo C, Gormley AK, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I[J]. Circ Res,2006,98: 63-72.
[11] Zhang Y, Ahmed AM, McFarlane N, et al. Regulation of SR-BI-mediated selective lipid uptake in Chinese hamster ovary-derived cells by protein kinase signaling pathways[J]. J Lipid Res,2007,48:405-416.
[12] Zhu W, Saddar S, Seetharam D, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity[J]. Circ Res,2008,102: 480-487.
[13] Iiyama K, Hajra L, Iiyama M, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation[J]. Circ Res,1999,85:199-207.
[14] Walpola PL, Gotlieb AI, Cybulsky MI, et al. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress[J]. Arterioscler Thromb Vasc Biol,1995,15:2-10.
[15] Murphy AJ, Woollard KJ, Hoang A, et al. High-density lipoprotein reduces the human monocyte inflammatory response[J]. Arterioscler Thromb Vasc Biol,2008,28:2071-2077.
[16] Smythies LE, White CR, Maheshwari A, et al. Apolipoprotein A-I mimetic 4F alters the function of human monocyte-derived macrophages[J]. Am J Physiol Cell Physiol,2010,298: C1538-C1548.
[17] Trigatti B, Rayburn H, Vinals M, et al. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A 1999,96: 9322-9327.
[18] Al-Jarallah A, Igdoura F, Zhang Y, et al. The effect of pomegranate extract on coronary artery atherosclerosis in SR-BI/APOE double knockout mice. Atherosclerosis 2013,228:80-89.
[19] Braun A, Trigatti BL, Post MJ, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 2002; 90: 270-6.
[20] Karackattu SL, Trigatti B, Krieger M. Hepatic lipase deficiency delays atherosclerosis, myocardial infarction, and cardiac dysfunction and extends lifespan in SR-BI/ apolipoprotein E double knockout mice. Arterioscler Thromb Vasc Biol 2006; 26: 548-54.
[21] Zhang S, Picard MH, Vasile E, et al. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation 2005; 111: 3457-64.
[22] Nakagawa-Toyama Y, Zhang S, Krieger M. Dietary manipulation and social isolation alter disease progression in a murine model of coronary heart disease[J]. PLoS One, 2012,7: e47965.
[23] Fuller M, Dadoo O, Serkis V, et al. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/ low-density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol,2014,34: 2394-2403.
[24] Covey SD, Krieger M, Wang W, et al. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells[J]. Arterioscler Thromb Vasc Biol,2003,23: 1589-1594.
[25] Braun A, Zhang S, Miettinen HE, et al. Probucol prevents early coronary heart disease and death in the high- density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A 2003; 100: 7283-8.
[26] Pei Y, Chen X, Aboutouk D, et al. SR-BI in bone marrow derived cells protects mice from diet induced coronary artery atherosclerosis and myocardial infarction. PLoS One 2013; 8: e72492.
[27] Yuhanna IS, Zhu Y, Cox BE, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase[J]. Nat Med,2001,7:853-857.
[28] Kocher O, Yesilaltay A, Cirovic C, et al. Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism[J]. J Biol Chem, 2003,278:52820-52825.
[29] Kocher O, Yesilaltay A, Shen CH, et al. Influence of PDZK1 on lipoprotein metabolism and atherosclerosis[J]. Biochim Biophys Acta,2008,1782:310-316.
[30] Yesilaltay A, Kocher O, Pal R, et al. PDZK1 is required for maintaining hepatic scavenger receptor, class B, type I (SR-BI) steady state levels but not its surface localization or function[J]. J Biol Chem,2006,281:28975-28980.
[31] Fernandez-Hernando C, Ackah E, Yu J, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease[J]. Cell Metab,2007,6:446-457.
[32] Kuhlencordt PJ, Gyurko R, Han F, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice[J]. Circulation,2001; 104:448-454.
[33] Yesilaltay A, Daniels K, Pal R, et al. Loss of PDZK1 causes coronary artery occlusion and myocardial infarction in Paigen diet-fed apolipoprotein E deficient mice[J]. PLoS One,2009,4:e8103.
[34] Mineo C, Yuhanna IS, Quon MJ, et al. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases[J]. J Biol Chem, 2003,278: 9142-9149.
[35] Cozen AE, Moriwaki H, Kremen M, et al. Macrophagetargeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death[J]. Circulation,2004,109: 2129-2135.
[36] Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice[J]. Circ Res,2004,95:637-644.
[37] Caligiuri G, Levy B, Pernow J, et al. Myocardial infarction mediated by endothelin receptor signaling in hypercholesterolemic mice[J]. Proc Natl Acad Sci U S A,1999,96:6920-6924.
✉ Bernardo L Trigatti, Department of Biochemistry and Biomedical Sciences, McMaster University and Thrombosis and Atherosclerosis Research Institute, McMaster University and Hamilton Health Sciences. David Braley Research Institute, Hamilton General Hospital Campus, 237 Barton St
E. Hamilton, Ontario L8L 2X2, Canada. Tel/Fax:(905) 521-2100 Ext. 40744/(905) 577-1427, E-mail: trigatt@mcmaster.ca.
28 May 2015, Accepted 29 June 2015, Epub 20 August 2015
R714.252, Document code: A
The authors reported no conflict of interests.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Organ donation in China: the major progress and the continuing problem
- Cerebral ischemia during surgery: an overview
- Apolipoprotein A-V gene therapy for disease prevention / treatment: a critical analysis
- Prevalence and risk factors of HIV and syphilis, and knowledge and risk behaviors related to HIV/AIDS among men who have sex with men in Chongqing, China
- Effects of closing and reopening live poultry markets on the epidemic of human infection with avian influenza A virus
- Sulindac sulfide selectively increases sensitivity of ABCC1 expressing tumor cells to doxorubicin and glutathione depletion