液相色谱-质谱联用法检测鸡蛋中8种氟喹诺酮类药物残留*
2016-12-02杨朝琳黄方取洪华嫦周鸿艳
杨朝琳,黄方取,洪华嫦,周鸿艳
(1.浙江师范大学 地理与环境科学学院,浙江 金华 321004;2.金华市疾病预防控制中心,浙江 金华 321002)
液相色谱-质谱联用法检测鸡蛋中8种氟喹诺酮类药物残留*
杨朝琳1,黄方取2,洪华嫦1,周鸿艳2
(1.浙江师范大学 地理与环境科学学院,浙江 金华 321004;2.金华市疾病预防控制中心,浙江 金华 321002)
建立了一种液相色谱-质谱联用法(LC-MS/MS)快速测定鸡蛋中8种氟喹诺酮类药物残留的方法.样品经磷酸-乙腈提取,冷冻离心,旋转蒸发浓缩,初始流动相溶解,然后用正己烷脱脂,有效地去除杂质,随后进行LC-MS/MS定性定量分析.8种氟喹诺酮类药物含量在2~50 μg/kg线性关系良好,相关系数R均大于0.995 6;添加量在5,10和20 μg/kg时,该方法的加标回收率为65.4%~119.6%,相对标准偏差均小于7.52%.该方法的检出限为0.000 5~0.22 μg/kg,定量限为0.02~0.74 μg/kg,适用于鸡蛋中8种氟喹诺酮类药物残留的定性和定量检测.
液相色谱-质谱联用;鸡蛋;氟喹诺酮;残留
沙拉沙星(Sarafloxacin)、二氟沙星(Difloxacin)、环丙沙星(Ciprofloxacin)、恩诺沙星(Enrofloxacin)和培氟沙星(Pefloxacin)等属于氟喹诺酮类药物,抗菌谱广、抗菌活性强,低毒、组织穿透力强,适用于预防和治疗呼吸、消化、泌尿系统的细菌感染症状,目前已被广泛应用于人类、家禽和水产养殖中.此类药物在体内消除缓慢,在家禽和水产饲养过程中该类药物的不合理使用会导致其残留在动物源性食品中,从而危害人类的健康.欧盟[1]、联合国粮农组织和世界卫生组织下的食品添加剂联合专家委员会(JECFA)[2]和我国[3]对动物源性食品中氟喹诺酮类药物残留量皆有规定.欧盟规定了氟喹诺酮类药物在鸡肉、动物肝脏和肾脏中的最高残留限量,如:恩诺沙星和环丙沙星为30 μg/kg,沙拉沙星为10 μg/kg[1].目前,动物源性食品中氟喹诺酮类药物残留问题已引起广泛的关注,开展动物源性食品中此类药物残留情况的监控,保障食品安全已是当务之急.
动物源性食品中氟喹诺酮类药物残留的检测方法,国家标准有液相色谱法(HPLC)[4]和高效液相色谱-质谱联用法(LC-MS/MS)[5];文献报道也较多,有免疫法[6]、薄层色谱法[7]、高效毛细管电泳法[8]、高效液相色谱法[9]和高效液相色谱-质谱联用法[10].高效液相色谱法是目前应用比较普遍的检测方法,常结合紫外检测器(UV)[11]、二极管阵列检测器(PDA)[12]和荧光检测器(FL)[13]使用,但荧光或紫外检测器的抗干扰能力相对较弱,对样品前处理和色谱分离条件要求高,分析时间较长,能同时检测的氟喹诺酮类药物种类较少,不能进行定性检测,要满足一些药物残留限量的检测要求,尚需提高灵敏度和可信度.高效液相色谱-质谱联用法的抗干扰能力强,可一次性检测多种氟喹诺酮类药物,灵敏度高、准确性好.
动物源性食品中杂质较多,样品前处理是检测氟喹诺酮类药物过程中一个重要的步骤,包括提取过程和纯化过程.动物源性食品中氟喹诺酮类药物残留检测多采用有机溶剂提取法,常用的提取纯化方法有固相萃取法[14]、液液萃取法[15]、超临界流体萃取法[16]等,也有微波辅助提取法[17]、加速溶剂萃取法[18]等较为创新的提取纯化方法.如:文献[19]用1%乙酸乙腈溶液为萃取液,采用微波萃取技术进行样品的前处理,经乙腈饱和的正己烷液-液脱脂纯化;文献[20]采用磷酸盐水溶液提取,HLB固相萃取柱纯化;文献[21]采用磷酸盐缓冲溶液和乙腈的混和溶液提取,提取液经正己烷液-液分配(LLP)除去脂肪后,用C18固相萃取(SPE)柱纯化.这些方法处理可得到良好的纯化效果,但操作复杂、实验成本高.
本研究采用反相液相色谱-质谱联用法,同时检测鸡蛋中8种氟喹诺酮类药物残留,无需进行固相萃取就可达到较好的纯化效果,与国家标准[4]和文献[22]报道的样品前处理方法相比,实验成本低、操作简单,方法的检出限、定量限更低,灵敏度提高,选择性和重现性好,能满足动物源性食品中氟喹诺酮类药物残留检测分析的要求.
1 材料与方法
1.1 试剂与仪器
沙拉沙星、二氟沙星、环丙沙星、恩诺沙星、培氟沙星、达氟沙星、司帕沙星和氧氟沙星均购自德国Dr.Ehrenstorfer公司.甲醇、乙腈、正己烷(色谱纯,Honeywell公司);甲酸(色谱纯,ROE SCIENTIFIC INC化学公司);实验用水为超纯水;盐酸(优级纯,浙江中星化工试剂有限公司);硫酸钠(纯度>99.9%,国药集团化学试剂有限公司).
Agilent 1290超高效液相色谱仪,6460三重四极杆质谱仪(美国Agilent公司);Milli-Q Advantage A10超纯水器(Millipore公司);WH-861涡流混合器(太仓市科教器材厂);Allegra 64R Centrifuge台式高速冷冻离心机(Beckman公司);R-215旋转蒸发仪(瑞士Buchi公司);N-EVAP氮吹仪(美国Organomation公司).
1.2 储备液和工作液的配制
1)单标准品储备液:称取8种氟喹诺酮类药物标准物质各10.0 mg,分别用甲醇溶解并定容至10 mL,得到1 000 μg/mL单标准品储备液,在-20 ℃储存备用.
2)混合标准品储备液:精确移取8种氟喹诺酮类药物的1 000 μg/mL单标准品储备液各10 μL于同一容量瓶中,以甲醇定容至1 mL,得10 μg/mL的混合标准品储备中间液.再取100 μL上述混合标准品储备中间液,用甲醇定容至1 mL,得1 μg/mL的混合标准品储备液,在-20 ℃储存备用.
3)混合标准品工作液:精确移取一定量的混合标准品储备液,用初始流动相定容,逐级稀释成50,20,10,5和2 μg/L的混合标准品工作液,于4 ℃储存备用.
1.3 样品的前处理
准确称取1.00 g鸡蛋试样(已匀质)于50 mL聚四氟乙烯离心管中,加入100 μL磷酸和10 mL乙腈,涡流混合60 s,超声提取10 min,以9 600 r/min离心10 min(-15 ℃),收集上清液于圆底烧瓶中;用10 mL乙腈重复提取一次;合并上清液,在45 ℃旋转蒸发至近干,剩余液体以氮气缓缓吹干,以1.0 mL初始流动相溶解残渣,用8 mL正己烷脱脂,重复3次,弃去正己烷层,下层清液以14 000 r/min离心5 min,上清液供高效液相色谱-质谱联用仪测定.
1.4 色谱和质谱条件
色谱柱:Eclipse plus C18柱(2.1 mm×100 mm,3.5 μm);柱温:30 ℃;样品室温度:室温;柱流速:0.30 mL/min;进样体积:10 μL;流动相A为0.2%甲酸水溶液,流动相B为乙腈/甲醇混合溶剂(V乙腈/V甲醇=50/50);梯度洗脱程序:0~1.0 min,5%流动相B;1.0~3.0 min,5%~20%流动相B;3.0~8.0 min,20%~40%流动相B;8.0~9.5 min,40%~98%流动相B;9.5~10.5 min,98%流动相B;10.5~11.5 min,98%~5%流动相B;11.5~14.5 min,5%流动相B.
质谱离子源:电喷雾离子源(ESI);扫描方式:正离子;检测方式:多反应监测(MRM);毛细管电压:3.50 kV;干燥气温度:350 ℃;干燥气流量:11 L/min;雾化气压力:3.1×105Pa;鞘气温度:380 ℃;鞘气流量:12 L/min;电子倍增器电压:200 V.其他参数见表1.图1为8种氟喹诺酮类药物的二级质谱图.
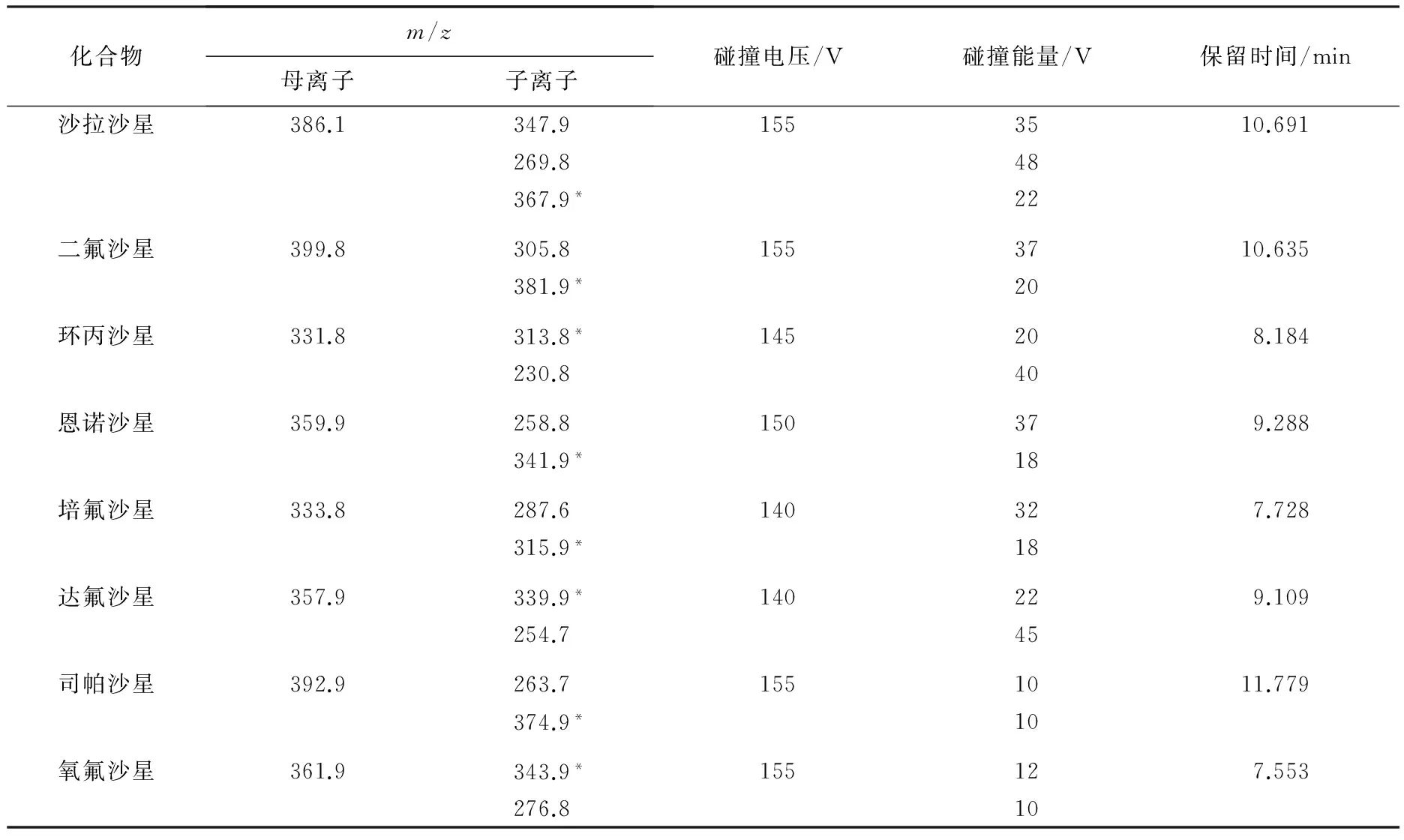
表1 8种氟喹诺酮类药物的质谱条件及相关参数
注:*代表定量离子.
运行开始时,色谱柱流出液经六通切换阀切换至废液中,质谱从4.0 min开始采集数据至12.5 min结束,六通切换阀又将色谱柱流出液切换至废液中.
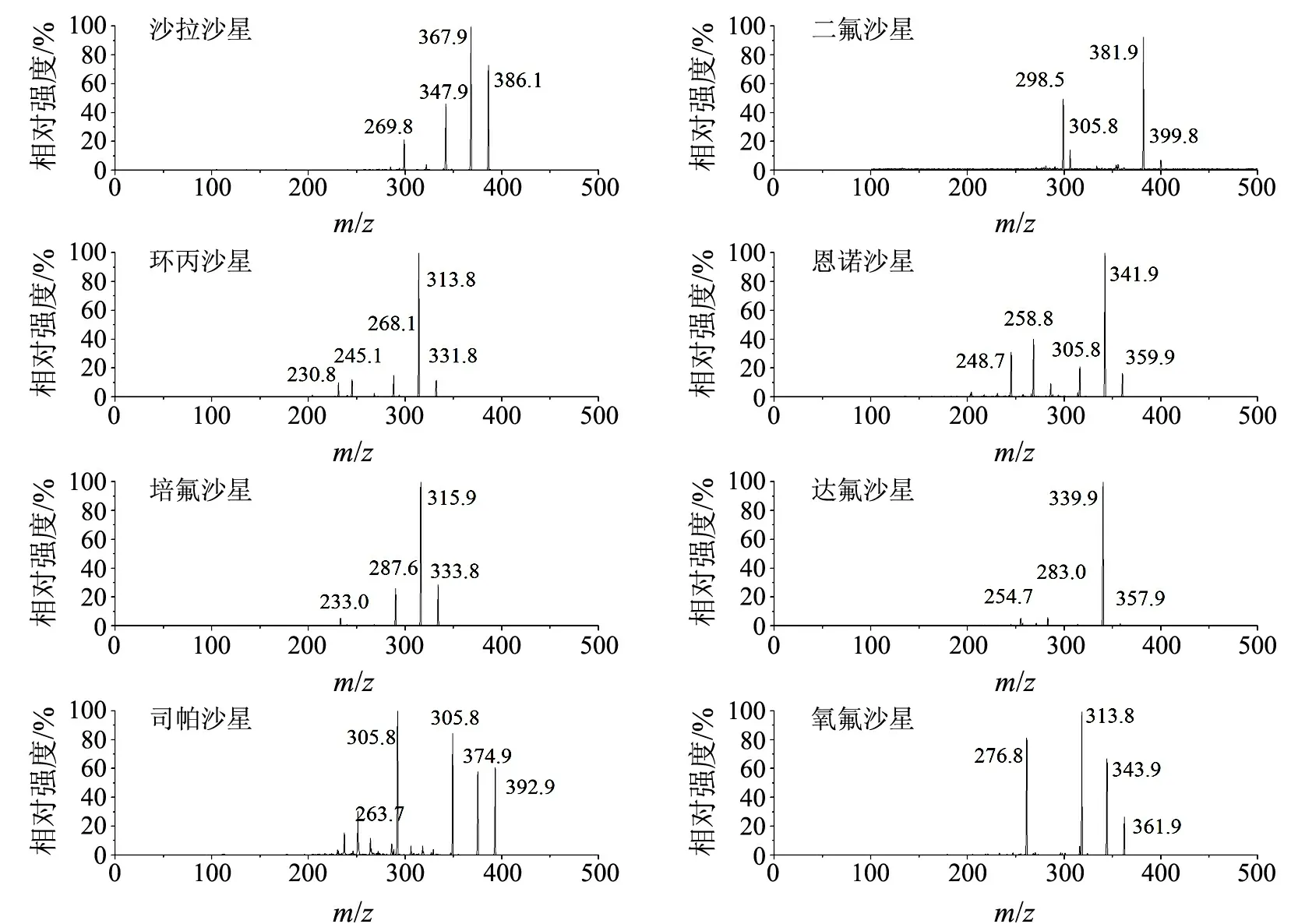
图1 8种氟喹诺酮类药物的二级质谱图
2 结果与讨论
2.1 方法的优化
2.1.1 提取条件优化
氟喹诺酮类药物残留的提取液一般为有机溶剂,常见的提取溶剂有酸化乙腈(V(盐酸)∶V(水)∶V(乙腈)=1∶1∶250)[23]、磷酸-乙腈[24]、1%乙酸-乙腈[25].本实验比较了用酸化乙腈、磷酸-乙腈和1%乙酸-乙腈3种溶剂进行提取时的回收率,结果如图2所示,以磷酸-乙腈的提取效率最高,酸化乙腈次之,1%乙酸-乙腈稍差.
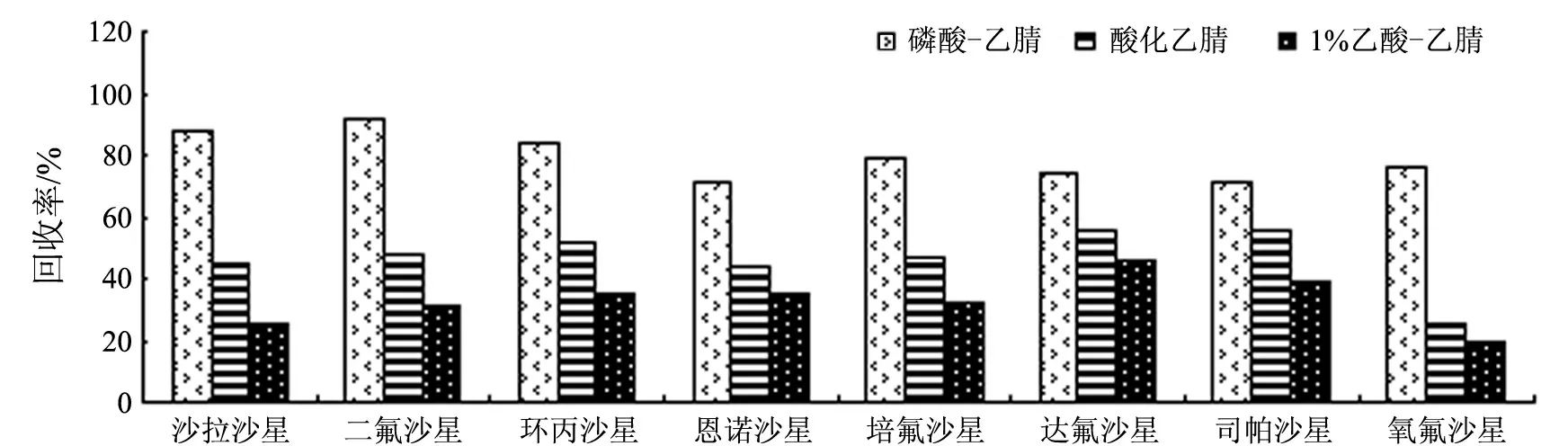
图2 提取液对8种氟喹诺酮类药物回收率的影响
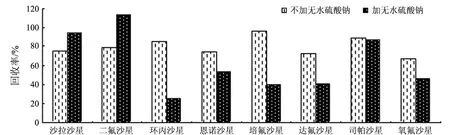
图3 无水硫酸钠对8种氟喹诺酮类药物回收率的影响
本实验也比较了样品提取时加无水硫酸钠与不加无水硫酸钠的提取效果,结果如图3所示,加入无水硫酸钠后环丙沙星、恩诺沙星、培氟沙星、达氟沙星和氧氟沙星的回收率降低,沙拉沙星和二氟沙星的回收率有所提高,对司帕沙星的回收率几乎不影响.考虑方法的简便性,本文选择不加无水硫酸钠的提取方法,即单独用磷酸-乙腈为提取液.
2.1.2 纯化条件优化
鸡蛋中油脂含量较高,如果处理不干净,会影响方法的准确度和结果的稳定性.乙腈提取液中含有许多共萃取物,以脂肪为主,采用高速冷冻离心辅以正己烷脱脂的除脂法,可以有效地去除样品中的脂肪类干扰物.在低温状态下,脂肪粒以固体状态从溶液中析出,未分离的油脂则被正己烷除去,以达到纯化分离的目的.本实验比较了常温离心(15 ℃)和冷冻离心(-15 ℃)对8种氟喹诺酮类药物回收率的影响,结果如图4所示,高速冷冻离心能提高环丙沙星、恩诺沙星、培氟沙星、达氟沙星和氧氟沙星的回收率,但对沙拉沙星、二氟沙星和司帕沙星的回收率影响不大.因此,本文选择高速冷冻离心辅以正己烷脱脂的除脂法.

图4 常温离心与冷冻离心对8种氟喹诺酮类药物回收率的影响
2.1.3 基质效应(ME)的消除
样品经磷酸-乙腈提取,然后冷冻离心辅以正己烷脱脂,得到良好的纯化,但仍无法完全消除基质效应.本实验采用提取后添加[26]的方法评价基质效应,比较了低浓度(5 μg/kg)和高浓度(20 μg/kg)下的基质效应,结果见表2.所测定的8种氟喹诺酮类药物均存在不同程度的基质增强效应,可能是样品、样品提取方法或纯化方法不同引起的.本文采用空白基质标准曲线,以消除基质效应对检测结果的影响.
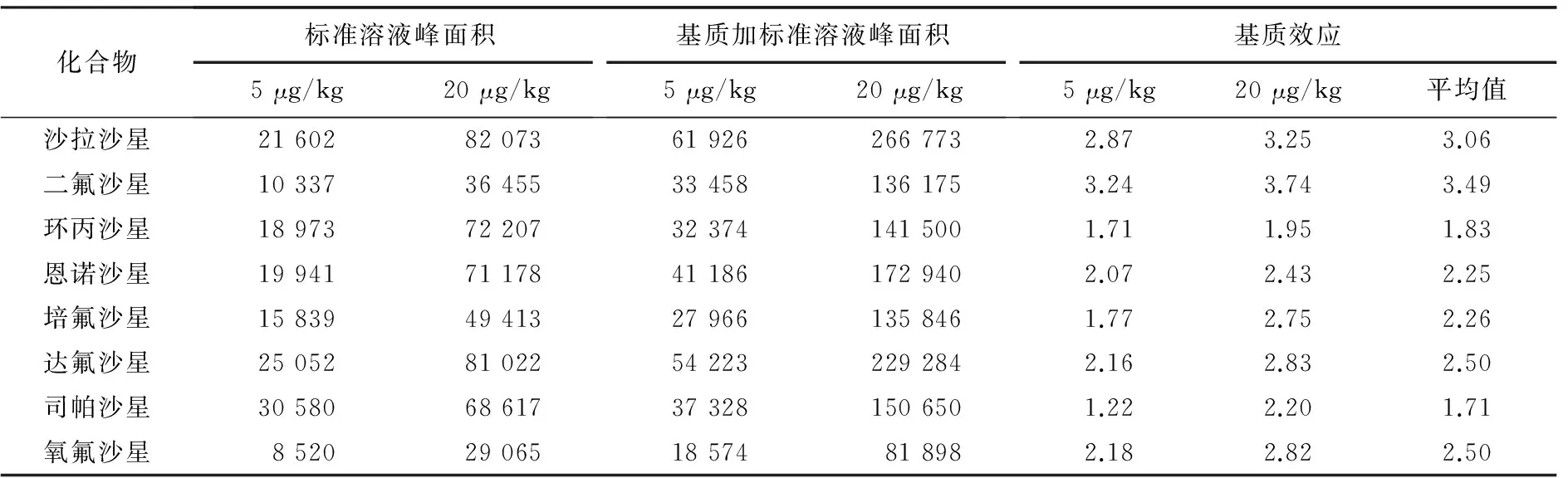
表2 8种氟喹诺酮类药物的基质效应
2.2 方法学论证
2.2.1 校正曲线和线性范围
称取1.00 g阴性鸡蛋样品,按上述方法处理样品,得到空白基质液,分别加入8种氟喹诺酮类药物混合标准溶液,配制成2,5,10,20和50 μg/kg的基质标准溶液.以所得8种氟喹诺酮类药物的离子峰面积平均值为纵坐标、标准溶液的浓度为横坐标,绘制标准曲线.表3为标准曲线回归方程和相关系数.由表3可见,8种氟喹诺酮类药物的质量分数在2~50 μg/kg时线性良好,线性相关系数R均大于0.995 6.
2.2.2 检出限和定量限
样品的检出限(LOD)是以基质标准曲线上的最低点(2 μg/kg),取信噪比S/N=3和样品处理过程的稀释倍数计算得出;样品的定量限(LOQ)是以基质标准曲线上的最低点(2 μg/kg),取信噪比S/N=10和样品处理过程的稀释倍数计算得出.如表3所示,8种氟喹诺酮类药物的方法检出限为0.000 5~0.22 μg/kg,定量限为0.02~0.74 μg/kg,达到了国家标准[4]和欧盟的要求,适用于大批量样品的快速筛选和定量测定.
2.2.3 准确度和精密度
取1.00 g阴性鸡蛋样品,进行5,10和20 μg/kg 3个水平的回收率实验.按照本文建立的方法处理样品,测定结果见表3.8种氟喹诺酮类药物的平均回收率为65.4%~119.6%.以所测各组分的峰面积计算其相对标准偏差(RSD,sr),衡量方法的精密度,8种氟喹诺酮类药物的RSD为0.33%~7.52%.因此,本方法的准确度和精密度均较高.
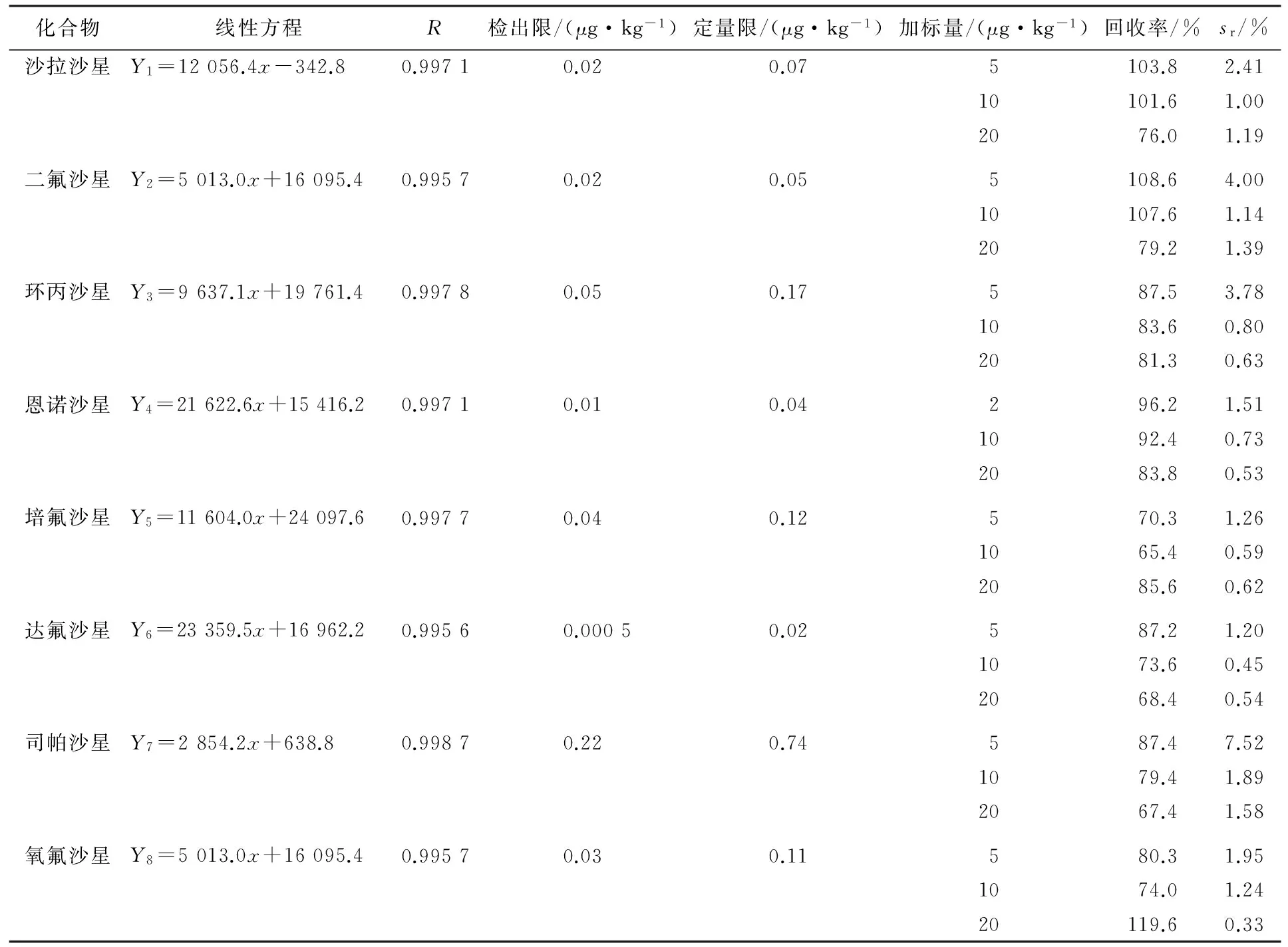
表3 8种氟喹诺酮类药物的线性方程、相关系数、检出限、定量限、回收率和精密度(n=6)
2.2.4 实样测定
对市场上采集的10份鸡蛋样品进行了测定,其中:2份样品检出氧氟沙星,含量分别为0.54和3.10 μg/kg;1份样品检出环丙沙星,含量为0.29 μg/kg.
3 结 论
本文建立了一种用LC-MS/MS快速测定鸡蛋中氟喹诺酮类药物残留的方法,能满足鸡蛋中8种氟喹诺酮类药物的检测要求.本方法不使用SPE小柱,并采用基质加标曲线消除基质效应,操作简单,经济,准确可靠,重复性好,灵敏度高.
[1]European Parliament and Council.Commission Regulation (EC) 508/1999 of 4 March 1999,a amending Annex I of Council Regulation (EEC) No 2377/90 of 26 June 1990 laying down a Community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin food[J].Official Journal of the European Communities,1999,60(2):16-17.
[2]World Health Organization.Evaluation of certain veterinary drug residues in food:Fiftieth report of the Joint FAO/WHO Expert Committee on Food Additives[R].Geneva:WHO,1999.
[3]中华人民共和国农业部.中华人民共和国农业部公告 第235号:动物性食品中兽药最高残留限量[J].中国兽药杂质,2003,37(2):7-9.
[4]中华人民共和国农业部,中华人民共和国国家卫生和计划生育委员会.GB 29692—2013 牛奶中喹诺酮类药物多残留的测定:高效液相色谱法[S].北京:中国标准出版社,2013.
[5]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.GB/T 21312—2007 动物源性食品中14种喹喏酮类药物残留检测方法:液相色谱-质谱/质谱法[S].北京:中国标准出版社,2008.
[6]Sliverlight J,Coldham N,Thorne L.Antibodies to the quinolones and fluoroquinolones for the development of generic and specific inmunossays for detection of these residues in animal products[J].Food Addit Contam,2003,20(3):221-328.
[7]Juhel-Gaugain M,Abjean J P.Screening of quinolone resvidues in pig muscle by planar chromatography[J].Chromatographia,1998,47(1/2):101-104.
[8]Juan-Garcia A,Font G,Pico Y.Determination of quinolones residues in chicken and fish by capillary electrophoresis[J].Electrophoresis,2006,27(11):2246-2249.
[9]Chung H H,Lee J B,Chung Y H,et al.Analysis of sulfonamide and quinolone antibiotic residues in Korean milk using microbial assays and high performance liquid chromatography[J].Food Chemistry,2009,113(1):297-301.
[10]郭伟,刘永,刘宁.超高效液相色谱-电喷雾串联质谱法同时分析鸡肉中7种氟喹诺酮类药物残留[J].色谱,2009,27(4):406-411.
[11]彭涛,雍炜,安娟,等.HPLC法同时测定牛奶中7种磺胺类药物和4种氟喹诺酮类兽药[J].中国卫生检验杂志,2011,21(1):353-355.
[12]饶钦雄,刘慧慧,刘向明,等.鱼组织中氟喹诺酮类药的HPCE多残留检测方法的建立[J].中国农学通报,2007,23(2):93-97.
[13]彭涛,雍炜,安娟,等.反相高效液相色谱法同时测定4种氟喹诺酮类药物在鸡可食性组织中的残留[J].色谱,2005,23(3):285-288.
[14]Herrera-Herrera A V,Hernández-Borges J,Rodríguez-Delgado M.Fluoroquinolone antibiotic determination in bovine,ovine and caprine milk using solid-phase extraction and high-performance liquid chromatography-fluorescence detection with ionic liquids as mobile phase additives[J].Journal of Chromatography A,2009,1216(43):7281-7287.
[15]潘媛,牛华,程晓云,等.高效液相色谱法检测六种氟喹诺酮类兽药残留前处理的优化[J].分析试验室,2011,30(5):69-72.
[16]于辉,赵萍.快速溶剂萃取-高效液相色谱-紫外串联荧光检测法测定太平湖白鱼中4种氟喹诺酮类药物残留[J].中国食品卫生杂志,2011,23(4):322-325.
[17]倪永付,朱莉萍,王勇,等.微波辅助萃取-液相色谱-串联质谱法检测猪肉中四种氟喹诺酮类药物残留[J].肉类工业,2012(9):42-44.
[18]厉文辉,史亚利,高立红,等.加速溶剂萃取-高效液相色谱-串联质谱法同时检测鱼肉中喹诺酮、磺胺与大环内酯类抗生素[J].分析测试学报,2010,29(10):987-992.
[19]刘靖靖,林黎明,江志刚,等.高效液相色谱法同时检测8种喹诺酮类兽药残留量[J].分析试验室,2007,26(8):5-9.
[20]赵思俊,李存,江海洋,等.高效液相色谱检测动物肌肉组织中7种喹诺酮类药物的残留[J].分析化学,2007,35(6):786-790.
[21]彭涛,雍炜,安娟,等.反相高效液相色谱/质谱法同时测定鸡肉中5种喹诺酮药物残留[J].分析化学,2006,34(S1):10-14.
[22]田媛,张尊建,李静,等.固相萃取-LC-MS/MS测定鸡蛋中氟喹诺酮类药物残留[J].中国药科大学学报,2010,41(1):60-65.
[23]中华人民共和国农业部.农业部783号公告—2—2006 水产品中诺氟沙星、盐酸环丙沙星、恩诺沙星残留量的测定 液相色谱法[S].北京:中国农业出版社,2007.
[24]林峰,林海丹,吴映璇,等.LC-MS-MS测定烤鳗中4种氟喹诺酮药物残留量[J].分析测试学报,2004,23(5):43-47.
[25]曹鹏,牟妍,高飞,等.分散固相萃取-超高效液相色谱-串联质谱法同时检测火锅食材中11种喹诺酮药物[J].色谱,2013,31(9):862-868.
[26]向平,沈敏,卓先义.液相色谱-质谱分析中的基质效应[J].分析测试学报,2009,31(9):753-756.
(责任编辑 薛 荣)
An improved method for determination of eight fluoroquinolones antibiotics in eggs by liquid chromatography tandem mass spectrometry
YANG Zhaolin1,HUANG Fangqu2,HONG Huachang1,ZHOU Hongyan2
(1.CollegeofGeographyandEnvironmentalSciences,ZhejiangNormalUniversity,Jinhua321004,China; 2.JinhuaMunicipalCenterforDiseaseControl&Prevention,Jinhua321002,China)
A method was established for the determination of eight fluoroquinolones antibiotics in egg sample by liquid chromatography tandem mass spectrometry.The sample was extracted with phosphoric acid and acetonitrile.After separated with high speed frozen centrifugation and concentrated with a rotary evaporator,the residue was dissolved with the mobile phase and then defatted withn-hexane.It was then analyzed with LC-MS.The linear ranges were from 2 to 50 μg/kg with the correlation coefficients above 0.995 6 μg/kg for all the 8 fluoroquinolones.The average recoveries of 8 fluoroquinolones were 65.4%~119.6% at three spiked level:5,10,20 μg/kg with the RSD lower than 7.52%.The limit of detection was of 0.000 5 μg/kg to 0.22 μg/kg.The limit of quantification was of 0.02 μg/kg to 0.74 μg/kg.The method could be applied to accurate qualitative and quantitative detection of the eight fluoroquinolones in the eggs.
liquid chromatography tandem mass spectrometry; egg; fluoroquinolone; residues
10.16218/j.issn.1001-5051.2016.01.010
��2015-04-08;
2015-10-22
国家自然科学基金资助项目(KYZKY11132);金华市科技局项目(2014-3-030)
杨朝琳(1991-),女,山东青岛人,硕士研究生.研究方向:地理环境与污染控制.通信作者:洪华嫦.E-mail:huachang2002@163.com
O657.7+2
A
1001-5051(2016)01-053-07
