超高效液相色谱串联质谱法同时测定马尾藻中4种内源性植物激素
2016-12-01林锡煌刘源森徐长安
易 勇 唐 旭 林锡煌 刘源森 林 凌 徐长安*
1(国家海洋局第三海洋研究所, 厦门 361005) 2(福建农林大学食品学院, 福州 350002)
超高效液相色谱串联质谱法同时测定马尾藻中4种内源性植物激素
易 勇1,2唐 旭1林锡煌1刘源森1林 凌1徐长安*1
1(国家海洋局第三海洋研究所, 厦门 361005)2(福建农林大学食品学院, 福州 350002)
建立了利用超高效液相串联质谱法(UPLC-MS/MS)同时测定马尾藻中4种植物激素(吲哚乙酸、吲哚丁酸、脱落酸、玉米素)的方法。马尾藻样品经过70%甲醇提取后,经PCX+PAX固相小柱净化,使用反相C18色谱柱分离。以5 mmol/L乙酸铵(含0.01%甲酸)和甲醇作为流动相进行梯度洗脱,采用多反应离子监测模式(MRM)分析测定。在0.01~1.0 μg/mL内,各种植物激素的相关系数均大于0.9990。4种植物激素的回收率为84.3%~102.1%,相对标准偏差为1.1%~6.4%。本方法的检出限为0.025~0.2 μg/kg。
马尾藻; 双柱联用; 超高效液相色谱串联质谱; 内源性植物激素
1 引 言
海藻是海洋资源的重要组成部分,是一种低等隐花植物,有绿藻、褐藻、红藻、蓝藻等10门。研究发现,海藻中含有许多活性物质,可以开发成海洋医药、保健、化妆用品等[1]。马尾藻是一类褐藻(Phaeophyta),作为海藻中的大型经济海藻,其种类超过250种,广泛分布于暖水和温暖的海域。我国的广东和广西沿海盛产马尾藻,约有60种[2]。大量研究发现,海藻中的促生长物质对陆生植物具有显著的促生长作用[3],已有一些基于海藻植物生长调节剂的商业产品出现[4]。我国作为农业大国,随着海藻生长调节剂的商业化进程,对于马尾藻植物激素的检测方法的开发显得十分的必要。
海藻植物激素是一类十分重要的内源植物激素,含有吲哚乙酸(IAA)、吲哚丁酸(IBA)、脱落酸(ABA)及玉米素(ZT)等植物激素[5,6]。目前,对于海藻中植物激素的研究报道很少,并且国内未建立相关标准,多组分植物生长调节剂检测方法的国家标准也尚未建立。因此选择合适的检测方法对于海藻植物激素的检测显得更加重要。目前,关于植物激素的检测方法主要有三大类,分别是生物测试、免疫检测和仪器分析方法。其中,仪器分析方法具有高灵敏度和高选择性的优势,特别是液相色谱-串联质谱法,是近年来生物化学中常用的检测技术[7~9]。
目前,还未见双柱联用净化方法用于马尾藻植物激素检测的报道,关于固相萃取文献中的方法大多数只能检测同一类性质的植物激素[10]。钟冬莲等[11]采用HPLC-MS/MS法成功测定了毛竹笋4种典型的内源性植物激素。本研究采用固相萃取双柱联用净化的方法,可以同时检测多组分不同性质的植物激素,大大缩短了检测时间。此外本研究采用超高效液相色谱串联质谱法对海藻中的4种植物激素同时进行分析检测,并探究了检测条件、前处理技术和方法学。
2 实验部分
2.1 仪器与试剂
Acquity超高效液相色谱仪(二极管列阵检测器,美国Waters公司); Extrapid柱-盘手动固相萃取仪(北京莱伯泰科仪器股份有限公司); QTrap5500线性离子阱串联质谱仪(美国AB Sciex公司); Milli-Q超纯水仪(美国Millipore公司); 循环水式多用真空泵(郑州长城科工贸有限公司); Neofuge 15R高速冷冻离心机(上海力申科学仪器有限公司); KQ-300E超声清洗器(昆山市超声仪器有限公司); BAS124S电子天平(赛多利斯科学仪器有限公司); R-215旋转蒸发器(德国BUCHI公司); 津腾溶剂过滤器(天津市津腾实验设备有限公司); IKA-Tisbasi组织匀浆机(德国IKA公司); 固相小柱PAX和PCX(月旭材料科技(上海)有限公司)。
标准品:吲哚乙酸、吲哚丁酸、玉米素、脱落酸 (美国Sigma公司); 甲醇(色谱纯,美国Spectrum公司); 甲酸(色谱纯,天津光复精细化工研究所); 乙酸铵(分析纯,国药集团化学试剂股份有限公司); 2,6-二叔丁基-4-甲基苯酚(BHT 生工生物工程(上海)股份有限公司); 氨水(分析纯,西陇化工股份有限公司); 水为Milli-Q超纯水; 缓冲溶液(含0.1%甲酸、0.5 mmol/L乙酸铵,pH 3.1)。
2.2 标准溶液的制备
标准溶液配制:分别准确称取10 mg(精确到0.01 mg)的IAA, IBA, ABA和ZT标准品,用100% 甲醇定容到100 mL,配制100 μg/mL的标准贮备液,密封, 于-20℃储存。分别吸取上述标准贮备液1 mL 于10 mL容量瓶中,用液相体系(60%缓冲溶液和40%甲醇,V/V)定容,配制成10 μg/mL的混合标准溶液,并进一步稀释成一系列的标准溶液,待测。
2.3 样品制备
2.3.1 样品前处理 马尾藻四月下旬采集于东山岛,洁净海水漂洗多次,用吸水纸将表面吸干,-20℃保存。参考QuEChERS(Quick Easy Cheap Effective Rugged Safe)快速前处理方法的称样量[12],准确称取10 g鲜海藻样品(精确至0.01 g),剪切破碎后,用匀浆机匀浆,加入30 mL 70% 甲醇溶液和5 mg抗氧化剂BHT,置于超声波中提取15 min。再将提取液于8000 r/min条件下离心5 min,上清液转移至100 mL烧杯中。将上清液于0.04 MPa、100 r/min、35℃条件下减压浓缩至1 mL。
2.3.2 样品净化
分别用2 mL 100% 甲醇活化和2 mL 0.05% 甲酸溶液平衡固相萃取小柱PCX,然后将前处理好的1 mL浓缩液上样,收集流出液; 再用2 mL 10% 甲醇溶液淋洗,收集淋洗液,最后用2 mL氨水-甲醇溶液(5∶95,V/V)洗脱,收集洗脱液。将淋洗液和流出液合并,上样于2 mL 100% 甲醇活化和2 mL 0.1% 甲酸平衡好的固相萃取小柱PAX,再用2 mL 10%甲醇溶液淋洗,最后用2 mL 甲酸-甲醇溶液(5∶95,V/V)洗脱,收集洗脱液。合并两次洗脱液,于0.03 MPa、100 r/min、35℃条件下减压浓缩至干。再准确加入1.0 mL配制好的缓冲水溶液涡旋振荡溶解残留物,经0.22 μm滤膜过滤后,使用UPLC-MS/MS分析检测。
2.4 超高效液相色谱串联质谱检测条件
2.4.1 超高效液相色谱条件 ACQUITY UPLC CSH C18色谱柱(100 mm×2.1 mm,1.7 μm; Waters公司); 柱温:35℃; 样品温度:25℃; 进样体积: 5 μL; 流速:0.3 mL/min; 流动相: 60% 缓冲溶液为流动相A、40%甲醇为流动相B; 洗脱程序:0~7 min,60%~20% A; 7~9 min,20%~60% A; 9~12 min,60% A。
2.4.2 质谱条件 离子源:电喷雾离子源(ESI); 扫描方式:多反应监测(MRM); 采用正离子扫描模式; 电喷雾电压(IS):5500 V; 雾化气电压(GS1):50 V; 碰撞气压力(CAD):Mediun; 气帘气电压(CUR):30 V; 辅助气电压(GS2):50 V; 离子源温度(TEM):500℃。
3 结果与讨论
3.1 前处理提取溶剂的优化
由于植物激素大多数是结构复杂的有机物[13],所以使用有机溶剂提取。本研究考察了不同浓度(100%, 90%, 70%和 50%)甲醇溶液的提取效果,为维持植物激素的稳定性,溶液中添加抗氧化剂BHT。每组设3个平行,通过UPLC-MS/MS法分别测定4种内源植物激素含量。
由表1可知,采用70%甲醇提取,得到的植物激素含量最高。由于植物激素吲哚乙酸在高温甚至常温下,时间过长易于分解; 如果水系过多,浓缩时需要更多的时间,不利于植物激素的提取,因此70%甲醇提取植物激素吲哚乙酸提取量较低。如果有机溶剂含量过多,有机溶剂除了溶解植物激素外,还溶解了植物体中含有的许多其它的有机物,它们之间可能发生物理化学反应,不利于吲哚乙酸的稳定,造成吲哚乙酸含量的减少,100%和90%甲醇提取吲哚乙酸含量较少可能与此有关。综合考虑,本研究选择70%甲醇进行马尾藻内源性植物激素的提取。
表1 不同提取方式所得马尾藻中4种植物激素的含量
Table 1 Plant hormone content with different extraction methods
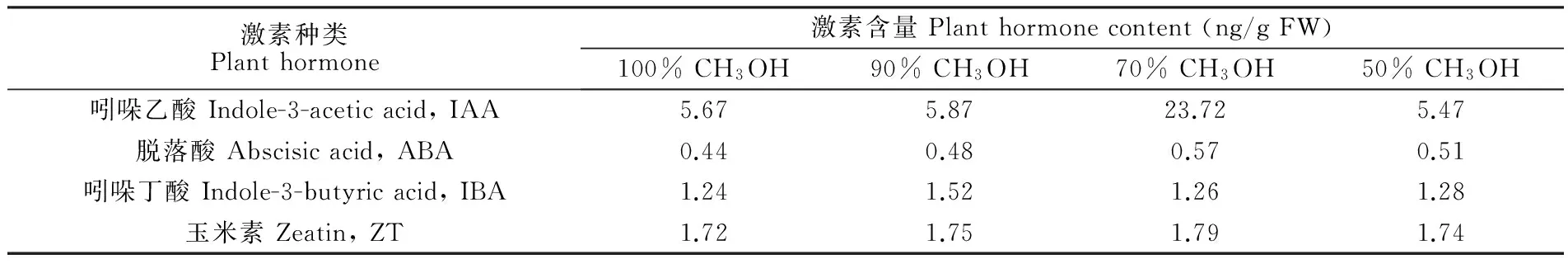
激素种类Planthormone激素含量Planthormonecontent(ng/gFW)100%CH3OH90%CH3OH70%CH3OH50%CH3OH吲哚乙酸Indole-3-aceticacid,IAA5.675.8723.725.47脱落酸Abscisicacid,ABA0.440.480.570.51吲哚丁酸Indole-3-butyricacid,IBA1.241.521.261.28玉米素Zeatin,ZT1.721.751.791.74
3.2 净化方法的优化
提取海藻植物激素常采用甲醇作为溶剂,然而在经多次萃取的离心后,会有大量色素、醌类和酚类等杂质溶入提取液中,如果这些物质不经过净化去除,就会对仪器的灵敏度和准确度产生很大影响,同时也会缩短色谱柱使用寿命。为了最大程度降低杂质的干扰,通常采用固相萃取小柱进行净化处理[14]。在植物激素中,同时含有酸性和碱性植物激素,所以采用单纯的阳离子或者阴离子固相小柱净化,都会使部分植物激素损失。在实验过程中,发现如果单独采用PAX固相小柱进行处理,采用10 μg/mL 混合标准品的测试,会发生部分损失,这样不利于植物激素的准确检测,其结果见图1a。由于PAX固相小柱为阴离子固相小柱,对阳离子(如H+)有吸附作用,对碱性的植物激素玉米素的吸附作用较差,其大部分作为流出液流出,检测结果见图1g,因此采用PCX+PAX双柱联用的方法。在优化相关条件基础上,双柱联用吸附效果较好(图1c)。
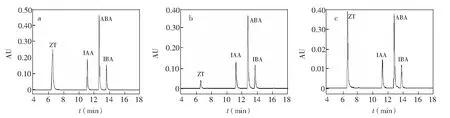
图1 4种植物激素混合标准品(a)及其经PAX固相小柱净化(b), 经PCX+PAX固相小柱净化(c) 的液相色谱图Fig.1 Chromatograms of the standard mixture (a), after purified by PAX solid-phase (b) and after purified by PCX and PAX solid-phase (c)色谱条件: ACQUITY UPLC CSH C18柱色谱柱(100 mm×2.1 mm,1.7 μm,Waters公司); 柱温: 35℃; 样品温度: 25℃; 进样体积: 10 μL; 流速: 0.15 mL/min; 波长268 nm; 流动相: 90%缓冲水溶液为流动相A、 10%甲醇为流动相B; 洗脱程序: 0~15 min,90%~10% A,15~18 min,10%~90% A,18~20 min,90% A。Conditions of (C): ACQUITY UPLC CSH C18 column (100 mm×2.1 mm,1.7 μm,Waters Co.); column temperature: 35℃; sample temperature 25℃; injection volume: 10 μL; flow rate: 0.15 mL/min; wavelenth: 268 nm; mobile phase: 90% buffer solution as mobile phase A; 10% methanol as mobile phase B; Elution program: 0~15 min,90%~10% A; 15~18 min,10%~90% A; 18~20 min,90% A.
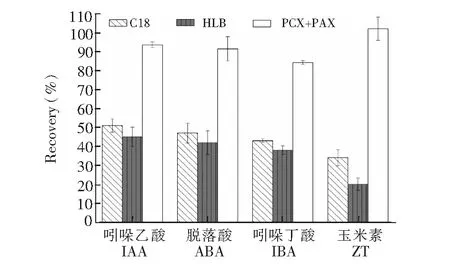
图2 3种固相萃取方式的净化回收率Fig.2 Recovery efficiencies for the three kinds of solid-phase extraction
固相萃取是植物激素研究常用的净化方式,为了考察净化效果和液相色谱柱上的保留行为,本研究考察了3种固相萃取方式,并以净化回收率作为考察依据,如图2所示,PCX+PAX的方式具有较好的回收率,回收率平均达到90%以上,而C18和HLB的回收率较差,平均不足50%,为了保证方法的净化效果和准确度,最终选择PCX+PAX双柱联用的方式。
3.3 色谱条件的优化
相对于BEH C18色谱柱,本研究选用的CSH C18色谱柱可以提供更好的峰形和峰之间的分辨率,并且杂质干扰较少[15]。
分别采用甲醇和乙腈作为流动相,发现流动相为乙腈时,样品出峰时间较早,当4种植物激素组分同时进样时,采用乙腈流动相标准品色谱峰之间的分辨率低于甲醇流动相的色谱标准品峰,因此采用甲醇作为洗脱流动相。同时考察了不同初始流动相比例,在初始流动相中甲醇体积占85%和10%,色谱峰的结果分别如图3所示,其它色谱条件见2.4.1节。
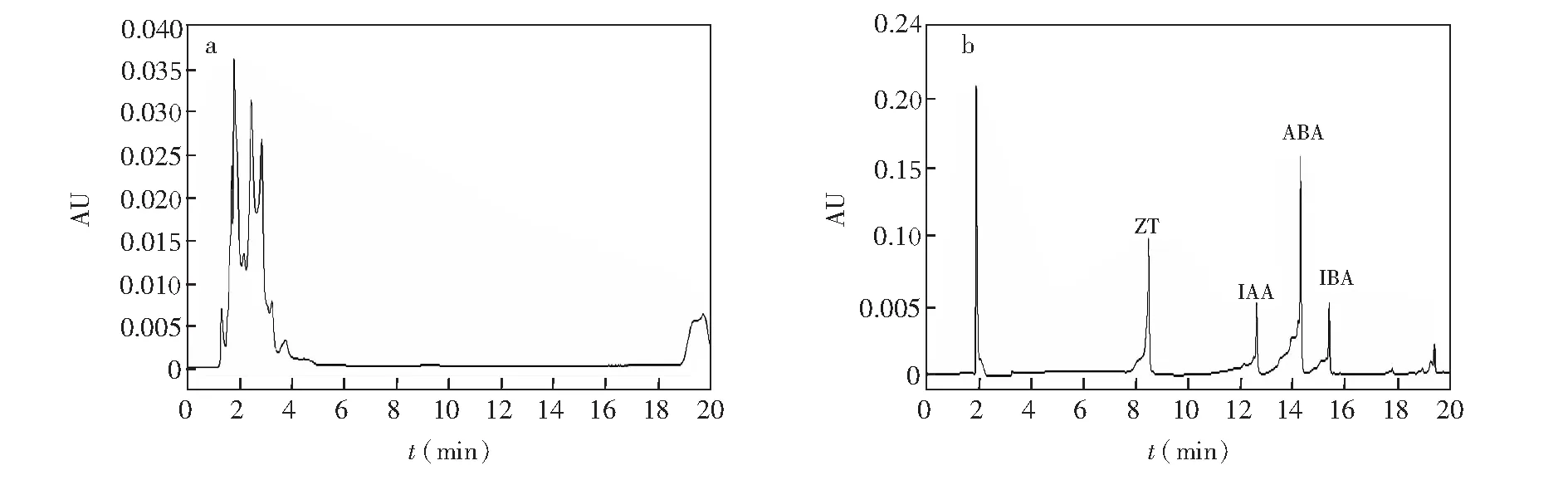
图3 采用85%甲醇初始流动相(a) 和10%甲醇初始流动相(b)的混合标准溶液色谱图Fig.3 Chromatograms of the mixed standard solution with 85% methanol of mobile phase (a) and with 10% methanol of mobile phase (b)
较高比例(85%)的甲醇不利于4种植物激素色谱峰的分开。可能是由于极性不够,不足以分开4种植物激素色谱峰。同时,当甲醇比例降低时,出现峰形较差和溶剂效应,在2 min左右出现溶剂峰。在本研究过程中发现, 在流动相的水系中添加少量乙酸铵可以增加离子化效率,进而表现出较好的峰形和灵敏度。此外,为了保证本实验的重复性和稳定性,同时维持水系酸度的稳定,在流动相的水系中再添加少量甲酸。图4为添加乙酸铵和甲酸的4种植物激素混合标准溶液的色谱峰图,检测器为Qtrap5500质谱检测器,检测条件见2.4.2节。
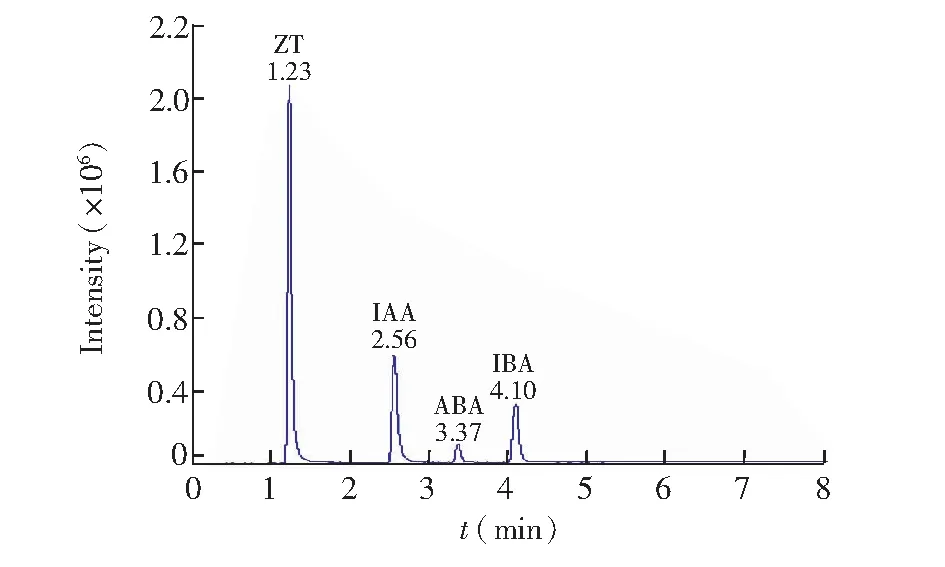
图4 添加乙酸铵和甲酸后四种植物激素混合标准溶液的色谱峰图Fig.4 Liquid chromatogram for the mixed standard solution of the four plant hormones with the addition of ammonium acetate and formate
3.4 质谱条件的优化
在电喷雾离子源(ESI)、正离子模式下,使用注射针泵,以20 μL/min的流速注入IAA、ABA、IBA和ZT标准品,在m/z100~300的扫描范围内分别对各物质进行MS onLy质谱扫描,确定IAA、ABA、IBA和ZT的分子离子,然后进行Product MS2扫描,调节碰撞能量CE(Collsion energy)确定合适的子离子(子离子质谱图如图5)。最后进行MRM模式扫描,分别对上述4种生长素优化CE 、去簇电压DP(Declustering potential)等,使信号强度达到最佳响应值(表2)。
3.5 方法评价
3.5.1 方法的标准曲线、线性范围和检出限 取10 μg/mL混合标准溶液, 配制成质量浓度分别为0.01, 0.1, 0.25, 0.5和1 μg/mL的混合标准溶液,绘制标准曲线(以质量浓度为X轴,对应的峰面积为Y轴)。结果表明,在0.01~1.0 μg/mL的质量浓度范围内具有良好的线性关系,相关系数均大于0.9990,见表3。
结果表明,本方法对4种化合物检出限较低,适于检测马尾藻内源性植物激素含量。另外,马尾藻通过净化后,4种植物激素的基质效应系数均为负值,具有明显的基质减弱效应,表明本方法中的前处理净化方式的净化效果明显,适用于马尾藻的基质。
表2 4种植物激素的质谱条件
Table 2 Mass spectrometry conditions for the four plant hormones

目标分析物Analyte母离子Parention(m/z)子离子Daughterion(m/z)碰撞能量Collsionenergy(eV)去簇电压Declusteringpotential(V)IAA176.0130.0*102.93543100IBA204.0130.1*144.1422195ABA265.1187.2*201.11917105ZT220.1136.0*118.9264890*定量离子(Quantitativeion)。
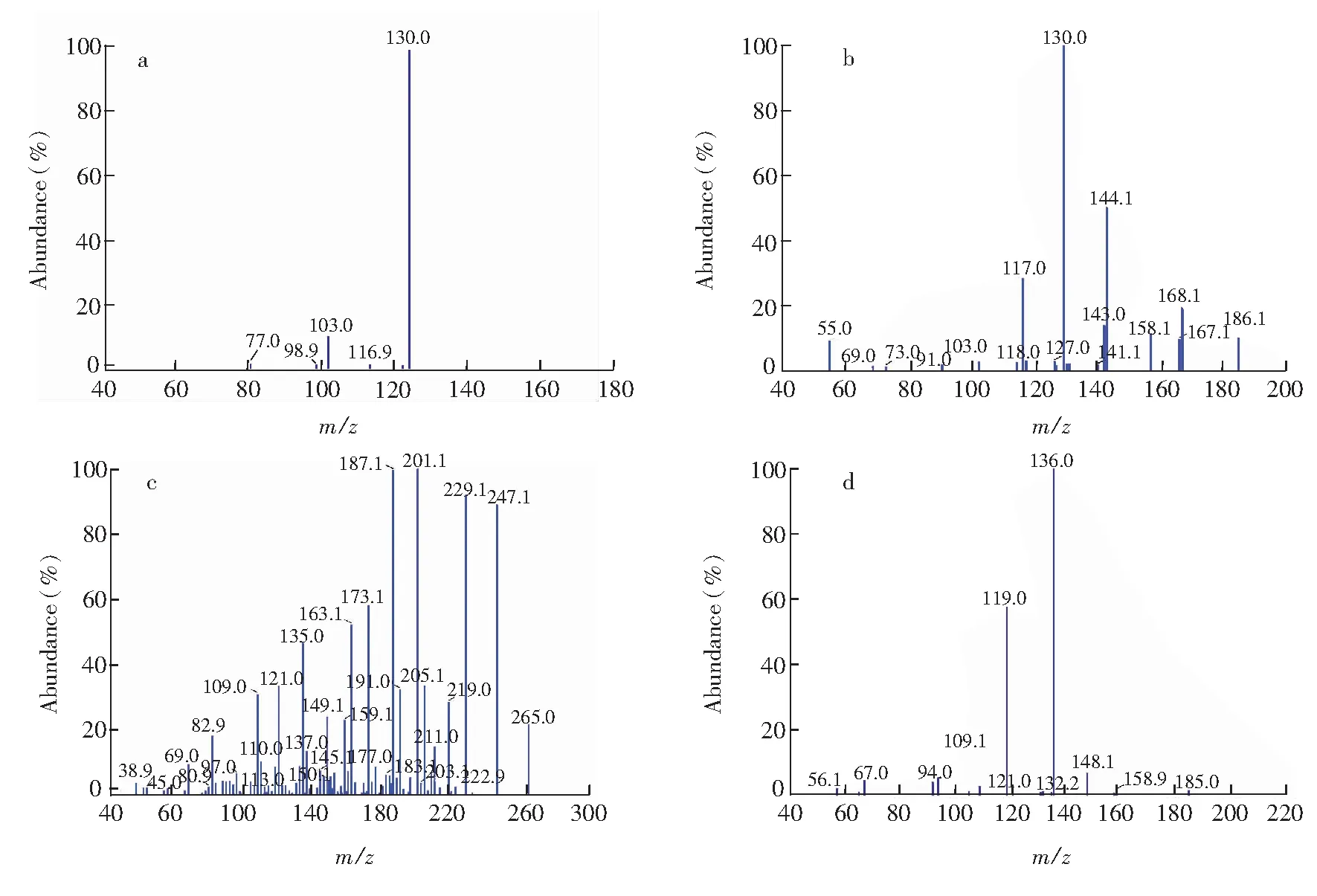
图5 IAA(a), IBA(b), ABA(c)和 ZT(d)的子离子质谱图Fig.5 Mass chromatograms of IAA ion (a), IBA ion (b), ABA ion (c) and ZT ion (d)
表3 4种植物激素的线性方程及检出限
Table 3 Linearity and detection limits of the four plant hormones

化合物Compound浓度范围Rangeofconcentration(μg/mL)线性方程Calibrationequation回归系数Regressioncoefficient(R2)基质效应系数*Matrixeffectts(η,%)检出限**Detectionlimit(μg/kg)IAA0.01~1y=9.51×106x+4.43×1040.9997-99.380.05IBA0.01~1y=4.84×106x-7.76×1030.9999-99.980.1ABA0.01~1y=3.51×107x+1.07×1050.9999-99.930.025ZT0.01~1y=1.12×108x-5.03×1040.9994-99.370.2*基质效应系数表示为η=(基质匹配标准曲线的斜率-溶剂匹配标准曲线的斜率)/溶剂匹配标准曲线的斜率;**按照信噪比S/N=3时,采用本方法检测出的植物激素的最低浓度即为检出限。*Thematrixeffectisexpressedascoefficientη=(theslopeofmatrix-matchedstandardcurve-theslopeofthestandardcurvematc-hingsolvent)/theslopeofthestandardcurvematchingsolvent;**WhentheratioofsignalovernoiseS/N=3,thelowestconcen-trationofplanthormonesdetectedbythismethodisdefinedasthedetectionlimit.
表4 不同样品激素的检测含量、回收率及相对标准偏差(n=3)
Table 4 Detected contents, recovery efficiencies and relative standard deviations for different hormone samples

样品激素Samplehormone检测含量*Detectioncontent*(μg/mL)回收率Recovery(%)相对标准偏差Relativestandarddeviation(%)IAA9.36±1.3993.61.5ABA9.16±3.3991.66.4IBA8.43±0.8984.31.1ZT10.21±3.59102.16.1*:平均值±SD,SD表示标准偏差。*:Mean±SD,SDstandsforstandarddeviation.
3.5.2 方法的准确度和精密度 分别向马尾藻的空白基质样品中添加10 μg/mL的混合标准溶液,按照2.3.2节的方法进行处理,通过对比峰面积计算回收率,见表4。
结果表明,本方法回收率较高,相对标准偏差小于10%。方法具有足够的灵敏性,准确可靠。
3.6 方法应用
为验证本方法的适用性和实用性,应用本方法对采自东山岛及市售的不同种类海藻(马尾藻、海带、龙须菜、海苔、红藻)进行检测,结果表明,样品中IAA植物激素含量均较高,1.59~15.93 μg/kg,远高于本方法的检出限; 其它植物激素(如IBA, ABA, ZT)含量较低,分别为0.57~3.11 μg/kg, 0.08~0.29 μg/kg和0~0.30 μg/kg。同时, 净化方法在基质效应方面可以进一步优化,为海藻中的植物激素检测分析提供一种可靠的方法。
4 结 论
本方法在样品前处理净化过程中,采用双柱联用净化的方法,同时净化并保留了酸性生长素和碱性生长素。使得可以同时检测马尾藻中的4种不同性质的植物激素含量。本方法是一种简单有效快捷的方法,满足对于马尾藻植物激素的检测需求,为海藻生长调节剂的商业化进程提供技术支持。
1 LÜ Hui-Min, ZHANG Kan.FoodScienceandTechnology, 1998, 6: 29-30
吕惠敏, 张 侃. 食品科技, 1998, 6: 29-30
2 ZHOU Qi-Cun, XIAO Feng-Bo.Marinescience, 2003, 27(3): 66-69
周歧存, 肖风波. 海洋科学, 2003, 27(3): 66-69
3 Gauraw K, Dinabandhu S.JournalofAppliedPhycology, 2011, 23(2): 251-255
4 QIN Qing, ZHANG Wen-Ju, ZHANG Tao.ChineseAgriculturalScienceBulletin, 2001, 17(1): 46-49
秦 青, 张文举, 张 涛. 中国农学通报, 2001, 17(1): 46-49
5 WANG Ze-Wen, SUN Wei-Hong, LIU Tao, ZHAI Yu-Xiu, XING Li-Hong, MIAO Jun-Kui, LENG Kai-Liang.ProgressinFisherySciences, 2011, 32(1): 94-98
王泽文, 孙伟红, 刘 涛, 瞿毓秀, 邢丽红, 苗钧魁, 冷凯良. 渔业科学进展, 2011, 32(1): 94-98
6 HAN Li-Jun, FAN Xiao, YUAN Zhao-Hui.OceanologiaEtLimnologiaSinica, 2003, 36(2): 167-170
韩丽君, 范 晓, 袁兆惠. 海洋与湖沼, 2003, 36(2): 167-170
7 TANG Li-Juan, WAN Yi-Qun.FoodScience, 2009, 30(21): 393-398
唐莉娟, 万益群. 食品科学, 2009, 30(21): 393-398
8 Xue J Y, Wang S L, You X W, Dong J, Han L, Liu F.RapidCommun.MassSpectrom., 2011, 25(21): 3289-3297
9 BAI Yu, DU Fu-You, BAI Yu, LIU Hu-Wei.ChineseBulletinofLifeSciences, 2010, 22(1): 36-44
白 玉, 杜甫佑, 白 玉, 刘虎威. 生命科学, 2010, 1(22): 36-44
10 Dobrev P I, Havlicek L, Vagner M, Malbeck J, Kaminek M.J.Chromatogr.A, 2005, 1075: 159-166
11 ZHONG Dong-Lian, DING Ming, TANG Fu-Bin, MO Run-Hong, TENG Ying.ChineseJ.Anal.Chem., 2013, 41(11): 1739-1743
钟冬莲, 丁 明, 汤富彬, 莫润宏, 滕 莹. 分析化学, 2013, 41(11): 1739-1743
12 Shi X M, Jin F, Huang Y T, Du X, Li C, Wang M, Shao H, Jin M, Wang J.J.Agric.FoodChem., 2012, 60(1): 60-65
13 Zhong Q S, Qiu X X, Lin C Y, Shen L, Huo Y, Zhan S, Yao J, Huang J, Kawano S, Hashi Y, Xiao L, Zhou T.J.Chromatogr.A, 2014, 1359: 131-139
14 HUANG Bing-Xin, FAN Xiao, HAN Li-Jun.OceanologiaEtLimnologiaSinica, 2002, 33(5): 509-514
黄冰心, 范 晓, 韩丽君. 海洋与湖沼, 2002, 33(5): 509-514
15 Flokova K, Tarkowska D, Miersch O, Strnad M, Wasternack C, Novak O.Phytochemistry, 2014, 105: 147-157
(Received 22 July 2015; accepted 18 September 2015)
Simultaneous Determination of 4 Kind of Plant Hormones in Sargassum by Ultra Performance Liquid Chromatography Tandem Mass Spectrometry
YI Yong1,2, TANG Xu1, LIN Xi-Huang1, LIU Yuan-Sen1, LIN Ling1, XU Chang-An*1
1(ThirdInstituteofOceanography,StateOceanicAdministration,Xiamen361005,China)2(CollegeofFoodScience,FujianAgricultureandForestryUniversity,Fuzhou350002,China)
A method of simultaneous determination of four kind of auxins (Indole-3-acetic acid, Indole-3-butyric acid, Abscisic acid, Zeatin) in sargassum by ultra performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) was established. After extracted by 70% methanol, the samples were enriched by the PCX and PAX solid-phase column. Then the samples were purified by reversed phase C18column using 5 mmol/L ammonium acetate (containing 0.01% formic acid) and methanol as a mobile phase in gradient elution. On the base of multiple reaction monitoring (MRM) analysis, the results showed that the correlation coefficient for all auxins examined exceeded 0.9990 in the range of 0.01-1.0 μg/mL. The recovery was from 84.3% to 102.1%, respectively. The relative standard deviation was less than 10%. Besides, the detection limit of this method was from 0.025 μg/kg to 0.2 μg/kg.
Sargassum; Double columns combination; Ultra performance liquid chromatography tandem mass spectrometry; Endogenous plant hormones
10.11895/j.issn.0253-3820.150585
本文系海洋公益性行业专项(No.201405038)、厦门南方海洋中心专项(No.14GZP004NF04)、国家海洋局第三海洋研究所基本科研业务费(海三科2013005)资助
2015-07-22收稿; 2015-09-18接受
* E-mail: xuchangan@tio.org.cn
