高脂饲养致小鼠胰岛素抵抗对脂肪肝形成的影响*
2016-11-24韦雪梅
韦雪梅, 邱 霓, 熊 燕
(广州医科大学药学院,广州蛇毒研究所,广东 广州 511436)
高脂饲养致小鼠胰岛素抵抗对脂肪肝形成的影响*
韦雪梅▲, 邱 霓▲, 熊 燕△
(广州医科大学药学院,广州蛇毒研究所,广东 广州 511436)
目的: 探讨高脂饲养致小鼠脂肪肝形成的机制。方法:随机将8周雄性C57BL/6J小鼠分成高脂饲养组(给予含60%卡路里的高饱和脂肪酸饲养)和正常对照组,饲养12周。监测体重、肝重、血甘油三酯、血总胆固醇、血糖和血胰岛素水平,通过高胰岛素正葡萄糖钳夹实验反映胰岛素敏感性,HE染色、苏丹IV染色及肝脂含量反映肝组织脂质沉积情况,确定高脂饲养致小鼠脂肪肝的形成。通过Western blot法检测磷酸化胰岛素受体底物1(IRS1)和蛋白激酶B(Akt)水平反映胰岛素信号通路激活情况,检测固醇调节元件结合蛋白1(SREBP-1)和脂肪酸合成酶(FAS)蛋白水平反映肝内脂质合成的情况。结果:高脂饲养组小鼠体重及肝重较正常对照组小鼠明显增加。与正常对照组相比,高脂组血和肝组织内甘油三酯和总胆固醇含量显著升高,血清胰岛素水平升高,葡萄糖输注率减少,磷酸化IRS1和Akt水平降低。肝组织HE染色可见高脂组肝细胞胞浆内充满大量脂肪空泡,苏丹IV染色可见肝细胞内存在大量大小不一的红色脂滴;SREBP-1和FAS蛋白水平明显升高。给予外源性油酸干预原代正常肝细胞48 h,磷酸化IRS1和Akt水平呈浓度依赖性减低,而SREBP-1和FAS蛋白表达明显升高。结论:高脂饲养导致小鼠肝脏发生胰岛素抵抗,并通过激活SREBP-FAS脂肪合成途径,促进肝脏脂质沉积,从而诱发脂肪肝。
胰岛素抵抗; 脂肪肝; 高脂饮食
随着全球肥胖人数的剧增,因肥胖所致的一系列代谢性疾病的发生率也随之增加[1]。饮食习惯的改变,大量高脂、高糖食物的摄入,导致营养过剩,是诱发肥胖的重要因素。胰岛素抵抗(insulin resis-tance,IR)是肥胖的主要特征之一,也是代谢性疾病如2 型糖尿病、心血管疾病、非酒精性脂肪肝等发展过程中重要的病理基础[2]。
肥胖患者脂肪组织内脂肪分解代谢活动旺盛,使细胞内甘油三酯(triglyceride,TG)分解释放游离脂肪酸(free fatty acids,FFA)增加,血液的游离脂肪酸水平升高,导致大量的游离脂肪酸进入肝脏和骨骼肌组织中,导致肝脏脂质沉积,促进脂肪肝的形成[3]。脂肪肝作为肝脏疾病的重要病理过程,如不及时治疗,可能加重并发生肝组织纤维化,甚至肝硬化。肝脏是胰岛素作用的主要靶器官之一;大量临床资料显示,几乎所有肥胖伴脂肪肝患者都存在周围组织和肝脏的胰岛素抵抗,且胰岛素抵抗的严重程度与脂肪肝的病情进展密切相关[4-5],但胰岛素抵抗促脂肪肝形成的分子机制仍未完全阐明,因此深入探讨胰岛素抵抗所致脂肪肝形成的分子机制,将有助于预防和治疗肥胖所致脂肪肝的形成。
本研究拟以高脂饲养致肥胖小鼠和经油酸处理的原代小鼠肝细胞为模型,观察高脂对肝脏组织及肝细胞胰岛素敏感性及脂质形成的影响,以探讨肥胖致胰岛素抵抗诱导脂肪肝形成的分子机制。
材 料 和 方 法
1 抗体和试剂
胰岛素受体底物1(insulin receptor substrate 1,IRS1)、p-Akt(Ser473)和Akt抗体购自Cell Signaling Technology;p-IRS1(Tyr632)抗体购自 Santa Cruz; HRP 标记的山羊抗兔 IgG、HRP 标记的山羊抗鼠 IgG、ECL 超敏发光液和BCA蛋白检测试剂盒购自碧云天公司;油酸(oleic acid,OA)购自Sigma。
2 实验方法
2.1 高脂饲养小鼠模型的建立 30只 8周龄雄性 C57BL/6J小鼠购自广东省医学实验动物中心,适应性饲养 1 周后,随机分为正常对照(control)组和高脂饲养(high-fat diet,HFD)组,分别给予10%卡路里普通饲料和60%卡路里的高脂饲料喂养12周,每周记录体重变化。实验用普通饲料和高脂饲料均购买于广东省实验动物中心。
2.2 高胰岛素正葡萄糖钳夹试验 高脂饲养第10周末,2组小鼠用10%的水合氯醛(按1 mL/300 g小鼠体重)经腹腔注射麻醉后,行颈静脉埋管术,待小鼠恢复1周。在第12周钳夹前1天晚给予小鼠禁食不禁水12 h;钳夹当天通过Harvard微量注射泵分别以25 mU/kg的初始浓度和4 mU·kg-1·min-1的维持浓度,持续120 min静脉泵入胰岛素,同时以30%葡萄糖液持续静脉泵入。每10 min通过鼠尾静脉测定1次血糖值,及时调节葡萄糖溶液的输注速度,以维持正常血糖水平。待血糖值稳定后,记录最后1 h内葡萄糖输注速率以评价胰岛素敏感性[6]。
2.3 血糖、血胰岛素、血脂和肝脏脂质的测定 雄性小鼠眼球取血,血液凝固后,以3 000 r/min,4 ℃离心10 min分离血清,-80 ℃保存。血清采用南京建成生物工程研究所的总胆固醇(total cholesterol,TC)和TG含量测定试剂盒进行检测。血清胰岛素按照武汉华美生物有限公司的小鼠胰岛素ELISA检测试剂盒进行检测。血糖采用电子感应法,用Roche的优越血糖仪测定鼠尾静脉血糖浓度。
小鼠处死后,迅速取出肝脏并置于液氮中,随后在-80 ℃保存。取出约30 mg肝脏组织,于300 mL生理盐水中剪碎并匀浆;12 000 r/min、4 ℃离心10 min后取上清到另一离心管内。用BCA法测定蛋白浓度并调整各样本至统一浓度。测量方法同血脂测定。
2.4 HE染色 小鼠颈椎脱臼法处死后迅速取出肝脏组织,称重后取部分肝组织以10%的甲醛溶液固定,制作常规石蜡切片,行HE染色,在Leica光学显微镜下进行观察并拍照。
2.5 苏丹Ⅳ染色 小鼠颈椎脱臼法处死后迅速取出部分肝脏组织包埋于OCT中,于冰冻切片机中进行连续切片,厚度为5 μm。将冰冻切片置于丙二醇中静止2 min后,置于苏丹Ⅳ染色缸内染色20 min;然后,将玻片分别置于85%乙醇、50%丙二醇中分化,用苏木精细胞核复染1 min,封片后在Leica光学显微镜下观察并拍照。
2.6 小鼠原代肝细胞的分离和培养 小鼠麻醉后,打开腹腔经下腔静脉插管,先以流速为5.5 mL/min灌注D-Hanks液(含10 mmol/L HEPES和0.5 mmol/L EDTA, pH 7.4, 37 ℃),待肝脏充分膨胀后,剪断门静脉;继续灌流3 min 后,改用含0.1%胰蛋白酶的D-Hanks液灌注,流速为4.5 mL/min;见包膜下肝组织呈龟背状裂开,取下肝脏,用4 ℃预冷PBS清洗肝脏,撕开包膜,制成肝细胞悬液。100目细胞筛过滤后,4 ℃、400 r/min离心5 min,弃上清,以含10% 胎牛血清的高糖DMEM重悬细胞,接种到6孔板中;5% CO2、37 ℃培养4 h后更换新鲜完全培养基,次日加药处理。
2.7 Western blot实验 取肝脏组织约30 mg,加入300 μL RIPA(含100 μmol/L PMSF),用弯眼科剪剪碎组织,使用机械匀浆机匀浆后,12 000 r/min、4 ℃离心10 min,取上清;采用BCA法测蛋白浓度。取30 μg蛋白进行SDS-PAGE,湿转法将蛋白条带转移到PVDF 膜上;用含5%脱脂奶粉封闭1 h 后依次加入 I 抗和 II 抗,洗膜后用ECL化学发光法显影,通过ChemiDoxTMXRS+凝胶成像系统采集图像,使用ImageJ软件进行灰度扫描。
3 统计学处理
利用SPSS 11.5 和Prism 5.0 统计软件进行数据处理和分析,所有数据均以均数±标准差(mean±SD)表示。所有数据进行正态性检验。两组样本均数比较采用两个独立样本的t检验,多组间均数比较用单因素方差分析,以P<0.05为差异有统计学意义。
结 果
1 高脂饲养对小鼠体重、肝脏重量的影响
饲养12周后,高脂饲养组小鼠体重明显超过正常对照组(P<0.01);高脂饲养组小鼠肝脏重量也较正常对照组增加(P<0.05)。与正常对照组相比,高脂饲养组小鼠的肝脏质量/体重比也明显增加(P<0.05),见表1。
表1 高脂饮食组和正常对照组小鼠体重、肝重及肝重/体重的比较
Table 1.The body weight, liver weight and ratio of liver weight to body weight in control group and HFD group (Mean±SD.n=10)

GroupBodyweight(g)BeforeAfterLiverweight(g)Liverweight/bodyweightControl22.5±1.730.6±1.11.25±0.200.041±0.006HFD22.3±1.337.2±1.6∗∗1.71±0.12∗0.048±0.002∗
*P<0.05,**P<0.01vscontrol group.
2 高脂饲养对小鼠葡萄糖、胰岛素及胰岛素敏感性的影响
虽经高脂饲养12周后,高脂饲养组和正常对照组小鼠空腹血糖浓度无显著差异,但高脂饲养组小鼠的血清胰岛素水平比正常对照组升高近2倍(P<0.01),见表2;同时,高胰岛素正葡萄糖钳夹实验结果表明,高脂饲养组小鼠平均葡萄糖输注率为(24.32±2.45) mg·kg-1·min-1,较正常对照组[(65.67±5.31) mg·kg-1·min-1]减低(P<0.01),说明为维持正常血清葡萄糖水平,在给予同等水平外源性胰岛素的情况下,高脂饲养组小鼠对外源性葡萄糖的处理能力仅为正常对照组的1/3水平(图1)。上述结果表明高脂饲养的肥胖小鼠已处于胰岛素抵抗阶段。
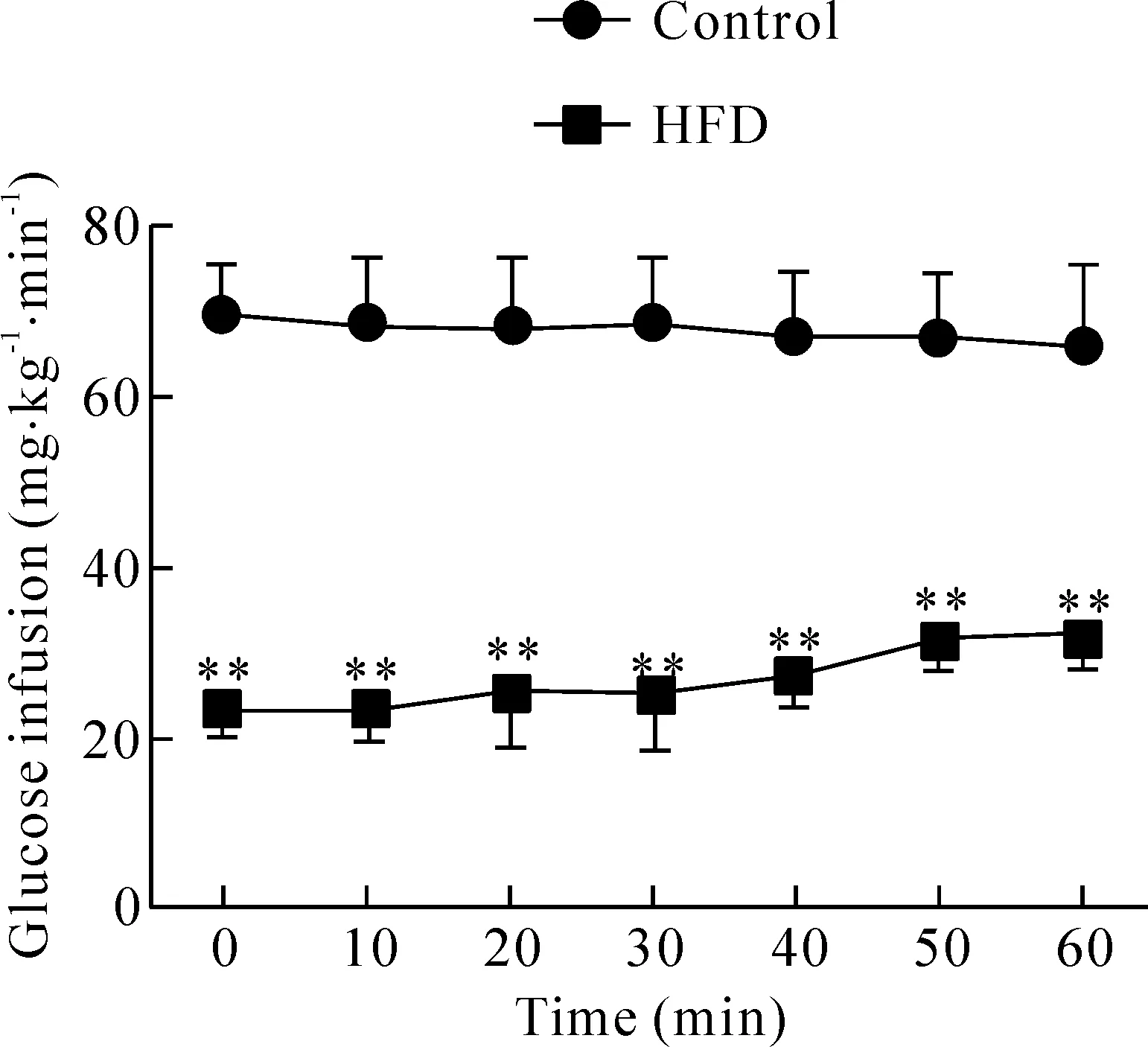
Figure 1.The result of hyperinsulinemic englycemic clamp experiment. Mean±SD.n=6.**P<0.01vscontrol.
图1 小鼠高胰岛素正葡萄糖钳夹实验结果
3 高脂饲养致肝脏组织形态学变化
正常对照组小鼠肝脏被膜光滑,呈红褐色,质地柔软;而高脂饲养组小鼠肝脏色泽较正常组暗淡,偏向于黄色,质地偏硬。HE染色可见高脂饲养组小鼠出现不同程度的弥漫性肝细胞脂肪变性,表现为肝细胞的胞浆内充满大量脂肪空泡。苏丹IV染色结果显示,对照组肝脏中未见明显的脂滴沉积,而高脂饲养组小鼠肝脏中存在大量大小不一的红色脂滴,进一步提示高脂导致小鼠肝脏内脂质沉积显著增多,见图2。

Figure 2.The HE staining (A) and Sudan IV staining (B) of the liver (×100).
图2 肝脏组织HE染色图和苏丹IV染色图
4 高脂饲养对小鼠血脂和肝脂的影响
高脂饲养显著增加小鼠血清甘油三酯和总胆固醇浓度(P<0.01),见表2。与病理形态学结果一致,高脂饲养组小鼠肝脏内甘油三酯和总胆固醇的含量明显高于正常对照组小鼠(P<0.05),见表3。
表2 高脂饲养组和正常对照组小鼠血清学相关指标的比较
Table 2.The serum biochemical indexes in the mice of control group and HFD group (Mean±SD.n=10)
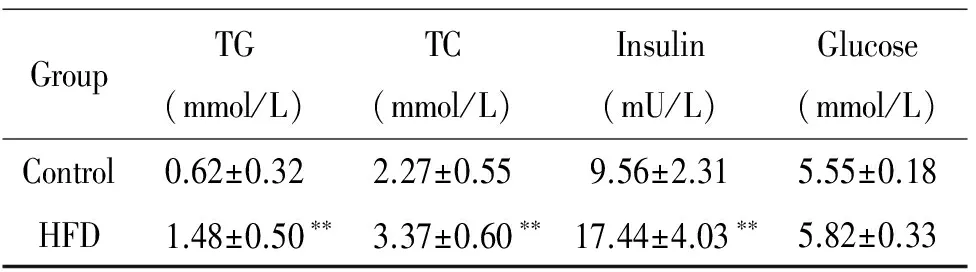
GroupTG(mmol/L)TC(mmol/L)Insulin(mU/L)Glucose(mmol/L)Control0.62±0.322.27±0.559.56±2.315.55±0.18HFD1.48±0.50∗∗3.37±0.60∗∗17.44±4.03∗∗5.82±0.33
**P<0.01vscontrol group.
表3 高脂饲养组和正常对照组小鼠肝脏脂肪相关指标的比较
Table 3.The contents of TG and TC in the liver of control group and HFD group (mmol/L. Mean±SD.n=10)
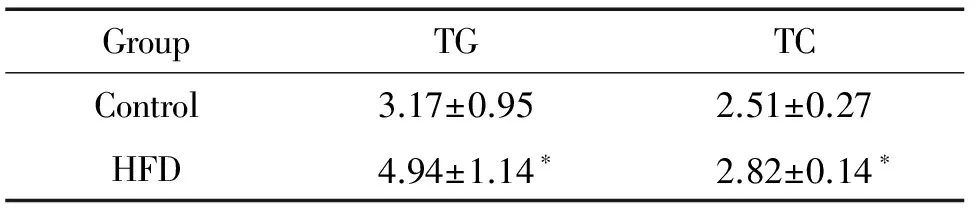
GroupTGTCControl3.17±0.952.51±0.27HFD4.94±1.14∗2.82±0.14∗
*P<0.05vscontrol group.
5 高脂饲养对小鼠肝脏组织胰岛素信号通路的影响
高脂饲养小鼠出现胰岛素抵抗的表现,肝脏作为胰岛素的靶器官之一,是否也存在胰岛素抵抗,我们随后检测了肝脏组织的胰岛素信号通路关键蛋白——IRS1和Akt的磷酸化水平。与正常对照组相比,高脂饲养组小鼠肝脏内IRS1的第632位酪氨酸磷酸化水平明显下调,表明高脂饲养小鼠肝脏对胰岛素的反应性降低,见图3。
6 高脂饲养对小鼠肝脏脂质合成的影响
为明确肝脏脂肪沉积的机制,我们检测了脂质合成相关蛋白的表达,结果表明,高脂饲养显著增加小鼠肝脏内脂肪酸从头合成的关键蛋白固醇调节元件结合蛋白 1(sterol regulatory element-binding protein-1,SREBP-1)及其下游蛋白脂肪酸合成酶(fatty acid synthase,FAS)的表达,见图4。
7 油酸促原代肝细胞胰岛素抵抗及脂质合成
为进一步明确高脂致胰岛素抵抗促脂肪肝形成的机制,我们采用酶消化法体外分离出8周正常雄性小鼠的肝脏细胞,并给予不同浓度油酸处理48 h。结果表明,随着油酸浓度的增加,IRS1磷酸化水平下调,其下游信号分子Akt的磷酸化水平也随之降低(图5),而肝细胞内SREBP-1和FAS的蛋白水平逐渐增加(图6)。
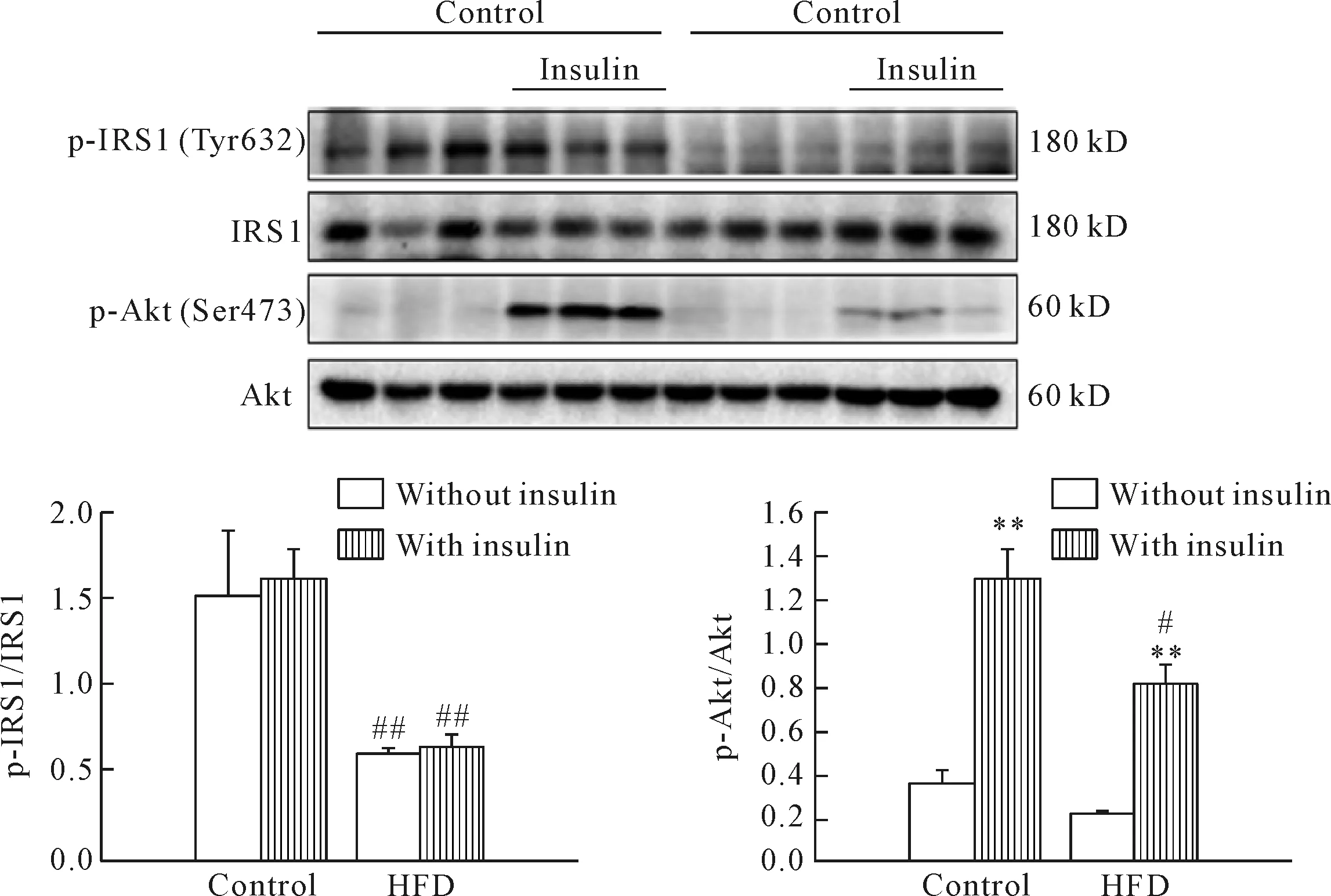
Figure 3.The expression of insulin signaling proteins in the liver of control group and HFD group. Mean±SD.n=6.**P<0.01vswithout insulin;#P<0.05,##P<0.01vscontrol group.
图3 肝脏组织胰岛素信号通路相关蛋白表达情况
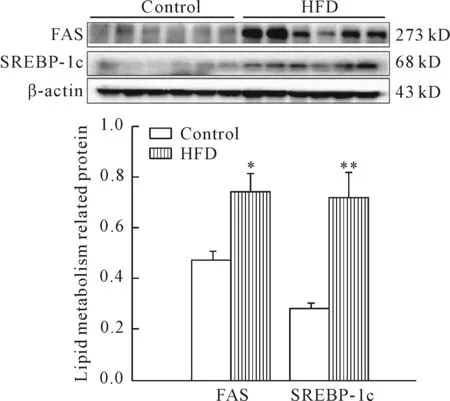
Figure 4.The expression of lipid synthesis proteins in the liver of control group and HFD group. Mean±SD.n=6.*P<0.05,**P<0.01vscontrol.
图4 肝脏组织脂质合成通路相关蛋白表达情况
Figure 5.The expression of insulin signaling proteins in the primary hepatocytes incubated with oleic acid for 48 h. Mean±SD.n=3.**P<0.01vscontrol.
图5 原代正常肝细胞油酸干预48 h后胰岛素信号通路相关蛋白变化情况
讨 论
本研究采用高饱和脂肪酸饮食饲养小鼠12周,发现小鼠体重及血脂水平高于正常对照组,提示高脂致肥胖小鼠模型建立成功;HE染色发现高脂饲养小鼠肝脏存在弥漫性肝细胞脂肪变性,表明高脂饲养致小鼠脂肪肝形成。目前高脂饮食致脂肪肝的机制仍不清楚。有研究报道,高脂饮食可导致胰岛素抵抗,表现为空腹胰岛素水平增加, 胰岛素敏感性降低[7-8]。我们也观察到高脂饲养小鼠的血胰岛素水平增加,机体对外源性葡萄糖的清除能力明显减弱,提示小鼠已存在胰岛素抵抗的表现;也进一步证实本研究通过高饱和脂肪酸饲养致胰岛素抵抗小鼠模型成功建立。
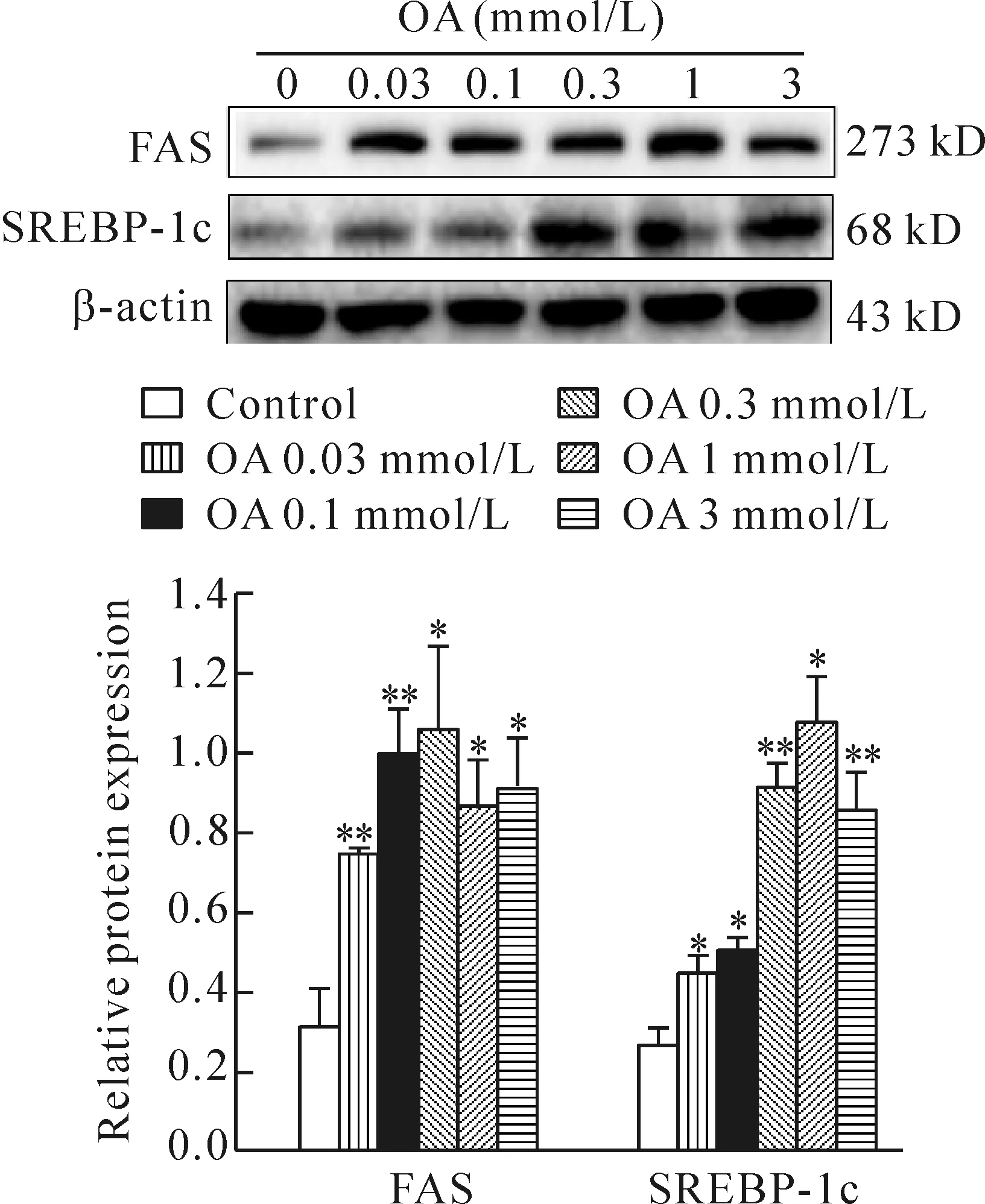
Figure 6.The expression of lipid synthesis proteins in the primary hepatocytes incubated with oleic acid for 48 h. Mean±SD.n= 3.*P<0.05,**P<0.01vscontrol.
图6 原代正常肝细胞油酸干预48 h后脂质合成相关蛋白变化情况
肥胖发生时,脂肪组织分解形成FFA的能力明显增加,导致经过门静脉输送到肝脏的FFA增加[9]。高FFA水平能干扰胰岛素在肝脏组织中与受体结合,显著减低胰岛素激活的IRS1酪氨酸磷酸化及下游信号分子Akt等活性,使胰岛素的生物效应降低,发生胰岛素抵抗[10]。本研究通过Western blot实验检测肝脏组织胰岛素信号通路相关蛋白发现,高脂饲养组小鼠肝脏组织的IRS1酪氨酸磷酸化水平和下游Akt丝氨酸磷酸化水平显著下降;另外,给予外源性油酸刺激原代肝细胞也发现,IRS1酪氨酸磷酸化水平和下游Akt丝氨酸磷酸化水平的活化呈剂量依耐性抑制,提示高脂不仅使小鼠发生全身胰岛素抵抗,还导致肝脏组织胰岛素信号转导受损,胰岛素敏感性降低。
在正常基础生理状态下,肝内脂质仅有 5%来源于内源性脂质从头合成途径;然而,在病理状态下如高脂环境,内源性脂质从头合成途径是肝内脂质沉积的一个重要来源[11]。SREBP是调节脂质从头合成酶类表达的最重要的上游转录因子,其下游靶基因编码的蛋白为肝脏脂质从头合成的3个下游关键酶:乙酰辅酶 A 羧化酶(acetyl-CoA carboxylase,ACC)、FAS和硬脂酰辅酶A脱饱和酶(stearyl-CoA desaturase,SCD)[13]。本实验观察到,高脂饮食增加小鼠肝脏SREBP-1 蛋白及其下游 FAS 蛋白表达,这结果也表明高脂致脂肪肝的形成主要与肝脏内脂质从头合成途径过度激活有关。一直以来,肥胖致肝脏脂质沉积和胰岛素抵抗之间的因果关系尚无定论;有研究者认为在脂肪肝形成过程中是先发生脂质的异常沉积再导致肝细胞发生胰岛素抵抗,即所谓的“脂毒性”;也有研究者认为是因为发生了胰岛素抵抗才导致脂质合成通路的异常激活,从而发生脂质的沉积[4, 10,13]。另外,IRS1/Akt信号通路对SREBP-1的作用在不同组织也存在差异。有报道证实,在HaCaT cells中,磷酸化的Akt可直接激活SREBP-1,促进脂肪酸合成[14];而在骨骼肌和肝脏中,磷酸化的Akt则主要通过激活腺苷酸活化蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)信号通路而抑制SREBP-1的活化[15-17]。在本研究中观察到,给予外源性油酸刺激原代肝细胞,导致IRS1/Akt胰岛素信号转导受抑制,在尚未出现明显肝细胞内脂肪沉积之前,SREBP-1 蛋白及 FAS 蛋白表达已明显升高,提示高脂饮食首先引起肝细胞内胰岛素信号通路受损,进而激活肝细胞内脂质从头合成信号通路,促使肝脏发生脂质沉积;但进一步的分子机制还有待后续深入研究。
综上所述,长期高脂饮食导致机体脂肪代谢紊乱,血脂含量增加,机体及肝脏组织对胰岛素敏感性下降,肝脏内脂质从头合成通路被激活,可能是导致肝脏组织内脂质大量沉积,脂肪肝形成的主要原因。
[1] Kahan S, Zvenyach T. Obesity as a disease: current policies and implications for the future[J]. Curr Obes Rep, 2016, 5(2):291-297.
[2] Shalitin S, Battelino T, Moreno LA, et al. Obesity, metabolic syndrome and nutrition nonalcoholic fatty liver di-sease: lipids and insulin resistance[J]. World Rev Nutr Diet, 2016, 114(2):21-49.
[3] Ress C, Kaser S. Mechanisms of intrahepatic triglyceride accumulation[J]. World J Gastroenterol, 2016, 22(4):1664-1673.
[4] Kanwar P, Kowdley KV. The metabolic syndrome and its influence on nonalcoholic steatohepatitis[J]. Clin Liver Dis, 2016, 20(2):225-243.
[5] Berk PD, Verna EC. Nonalcoholic fatty liver disease: lipids and insulin resistance[J]. Clin Liver Dis, 2016, 20(2):245-262.
[6] Declercq J, Kumar A, Van Diepen JA, et al. Increased beta-cell mass by islet transplantation and PLAG1 overexpression causes hyperinsulinemic normoglycemia and hepatic insulin resistance in mice[J]. Diabetes, 2010, 59(8):1957-1965.
[7] Coelho DF, Pereira-Lancha LO, Chaves DS, et al. Effect of high-fat diets on body composition, lipid metabolism and insulin sensitivity, and the role of exercise on these parameters[J]. Braz J Med Biol Res, 2011, 44(10):966-972.
[8] 任路平,宋光耀,章冬梅,等. 短期高果糖喂养对小鼠肝脏脂质沉积和胰岛素敏感性的影响[J]. 中国病理生理杂志, 2011, 27(12):2286-2290.
[9] Grundy SM. Adipose tissue and metabolic syndrome: too much, too little or neither[J]. Eur J Clin Invest, 2015, 45(11):1209-1217.
[10]Bugianesi E, Moscatiello S, Ciaravella MF, et al. Insulin resistance in nonalcoholic fatty liver disease[J]. Curr Pharm Des, 2010, 16(17):1941-1951.
[11]Lambert JE, Ramos-Roman MA, Browning JD, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease[J]. Gastroenterology, 2014, 146(3):726-735.
[12]Li J, Luo J, Xu H, et al. Fatty acid synthase promoter: characterization, and transcriptional regulation by sterol regulatory element binding protein-1 in goat mammary epithelial cells[J]. Gene, 2015, 561(1):157-164.
[13]Asrih M, Jornayvaz FR. Metabolic syndrome and nonalcoholic fatty liver disease: is insulin resistance the link?[J]. Mol Cell Endocrinol, 2015, 418(Pt 1):55-65.
[14]Zhou BR, Huang QH, Xu Y, et al. Dihydrotestosterone induces SREBP-1 expression and lipogenesis through the phosphoinositide 3-kinase/Akt pathway in HaCaT cells[J]. Lipids Health Dis, 2012, 11:156.
[15]Quan HY, Kim do Y, Kim SJ, et al. Betulinic acid alleviates non-alcoholic fatty liver by inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling pathway[J]. Biochem Pharmacol, 2013, 85(9):1330-1340.
[16]Seo MS, Kim JH, Kim HJ, et al. Honokiol activates the LKB1-AMPK signaling pathway and attenuates the lipid accumulation in hepatocytes[J]. Toxicol Appl Pharmacol, 2015, 284(2):113-124.
[17]Wu W, Tang S, Shi S, et al. Metformin attenuates palmitic acid-induced insulin resistance in L6 cells through the AMP-activated protein kinase/sterol regulatory element-binding protein-1c pathway[J]. Int J Mol Med, 2015, 35(6):1734-1740.
(责任编辑: 林白霜, 罗 森)
Effect of insulin resistance on fatty liver in high-fat diet-fed miceWEI Xue-mei, QIU Ni, XIONG Yan
AIM: To study the influence of insulin resistance on fatty liver in the mice fed with high-fat diet (HFD). METHODS: Male 8-week-old C57BL/6J mice were randomly divided into HFD group (with 60% calories by high saturated fatty acid) and control group (with chow diet).The mice in both groups were fed for 12 weeks. The body weight, liver weight, serum triglyceride (TG) and total cholesterol (TC), and blood glucose and insulin levels were measured. Hyperinsulinemic euglycemic clamp experiment was applied to reflect insulin sensitivity. The lipid deposition in the liver was analyzed by HE staining, Sudan IV staining and measurement of liver fat content. The phosphorylation levels of IRS1 and Akt, and the protein levels of SREBP-1 and FAS were determined by Western blot to reflect the activities of insulin signaling and lipid synthesis. RESULTS: Compared with control group, the body weight and liver weight were significantly increased in HFD group. TG and TC contents in serum and liver tissues were remarkably increased in HFD group. High-fat diet induced insulin resistance, as evidenced by increased serum insulin levels, reduced glucose infusion rate and decreases in IRS1 and Akt phosphorylation levels. In livers of HFD group, HE staining showed that the cytoplasm of hepatocytes was filled with vacuoles. Sudan IV staining also displayed that many different sizes of red lipid drops existed in the hepatocytes, and the protein levels of SREBP-1 and FAS were significantly increased. In primary normal hepatocytes with exogenous oleic acid intervention for 48 h, the phosphorylation levels of IRS1 and Akt were reduced, and the protein expression of SREBP-1 and FAS was significantly increased in a dose-dependent manner. CONCLUSION: Feeding with HFD leads to insulin resistance, resulting in activation of lipid synthesis and accumulation of lipid deposition in the liver, thus inducing fatty liver.
Insulin resistance; Fatty liver; High-fat diet
1000- 4718(2016)10- 1875- 06
2016- 04- 27
2016- 06- 20
中国博士后基金资助项目(No.2012M521590; No.2013T60792);广东省自然科学基金资金项目(No.s2013040014350)
△ 通讯作者 Tel: 020-37103273; E-mail: xiongyan2001@yahoo.com
▲ 并列第 1 作者
R363.2
A
10.3969/j.issn.1000- 4718.2016.10.022
杂志网址: http://www.cjpp.net
(GuangzhouInstituteofSnakeVenomResearchandSchoolofPharmaceuticalSciences,GuangzhouMedicalUniversity,Guangzhou511436,China.E-mail:xiongyan2001@yahoo.com)
