核壳结构催化剂应用于质子交换膜燃料电池氧还原的研究进展
2016-11-22骆明川蔡业政孙照楠
朱 红 骆明川 蔡业政 孙照楠
(北京化工大学理学院,化工资源有效利用国家重点实验室,北京100029)
核壳结构催化剂应用于质子交换膜燃料电池氧还原的研究进展
朱红*骆明川蔡业政孙照楠
(北京化工大学理学院,化工资源有效利用国家重点实验室,北京100029)
质子交换膜燃料电池(PEMFCs)由于高比功率密度、高能量转换效率、环境友好和低温下快速启动等优点受到广泛关注,被认为是替代传统内燃机成为汽车动力的最理想能源转换装置。目前PEMFCs仍需较高载量的贵金属Pt作为电催化剂以保持转换效率,因此,开发低Pt量高活性的电催化剂对PEMFCs技术的商业化进程至关重要。核壳结构催化剂被证明是一种能有效降低电极Pt用量的策略,其既能通过结构优势提高贵金属Pt的利用率,又能通过电子或几何效应改善催化剂的催化活性和稳定性。本文首先简介了PEMFCs阴极氧还原反应(ORR)电催化剂构效关系的理论研究;其次综述了几种典型核壳结构电催化剂应用于ORR的研究进展;最后对ORR低Pt电催化剂的下一步研究方向作了展望。
质子交换膜燃料电池;氧还原反应;低Pt催化剂;核壳结构;电子/几何效应

朱红,1957年生。1998年博士毕业于中国矿业大学化工学院。现为北京化工大学理学院,化工资源有效利用国家重点实验室,现代催化研究所所长。教授、博士生导师。主要从事燃料电池阴极催化剂,膜材料的设计与制备,能源化学等方面的基础研究。主持完成国家自然科学基金5项,多次担任国家863、973项目子课题负责人。

蔡业政,1989年生。2013年本科毕业于广西师范大学化学化工学院,2013至今就读北京化工大学理学院研究生。主要研究方向为质子交换膜燃料电池阴极核壳型铂基催化剂合成、表征及应用。参与国家自然科学基金1项。

骆明川,1987年生。2009年本科毕业于北京化工大学应用化学专业,2010年至今在北京化工大学理学院化学专业攻读博士。研究方向为低铂核壳结构电催化剂应用于质子交换膜燃料电池阴极氧还原反应。参与国家自然科学基金项目2项。

孙照楠,1989年生。2012年本科毕业于北京化工大学应用化学系,2012年至今就读于北京化工大学理学院化学专业硕博连读。主要研究方向为燃料电池阴极非贵金属催化剂合成、表征及应用。参与国家自然科学基金2项。
1 引言
为实现人类社会的可持续发展,摆脱化石燃料的束缚和开发利用可再生的清洁能源成为了这个世纪全球各国的迫切任务1。氢能具有储量丰富和环境友好两大优点,被认为是未来社会的理想能源形式。而燃料电池不经过卡诺循环的限制,能高效地将化学能转化为电能,被认为是利用氢能的最佳能源转换设备。其中,质子交换膜燃料电池(PEMFCs)是被研究最广的燃料电池类型,其能量密度高、低温快速启动、结构简单、安全性佳和噪音低等优点使其成为替代内燃机作为汽车动力电源的最佳装置2。2014年12月15日,日本丰田汽车公司正式发售世界第一款基于PEMFCs技术的氢燃料电池汽车Mirai(日文意思:未来),成为燃料电池技术应用于汽车动力电源的里程碑。其他国际著名汽车厂商如本田、现代、通用等也相继发行了各自旗下的燃料电池汽车,国内的上汽基团则在2015年的上海车展上展示了自主研发的荣威950插电式燃料电池汽车。
在实际应用中,PEMFCs通常需要使用高活性的电催化剂以降低电极反应(尤其是阴极反应)缓慢的动力学所引起的高过电势,而稀缺贵金属Pt依然是最常用且高效的电催化材料3。尽管目前使用的碳载高分散的纳米Pt催化剂(Pt/C)已大大地提高了Pt的利用率,但常见PEMFCs电极中的Pt用量仍维持在0.4 mg·cm-2左右,其中阳极氢氧化反应(HOR)的用量仅为0.05 mg·cm-2,而阴极氧还原反应(ORR)的用量则通常高达0.35 mg·cm-2。若要实现PEMFCs被大规模地应用为汽车的动力电源,则需要将其电极中的Pt用量降低至目前内燃机汽车尾气处理的Pt用量水平(目前家用汽车的平均用量为每辆车5 g Pt)。因此,设计开发低成本、高活性和长寿命的阴极ORR低Pt电催化剂一直是PEMFCs技术研究领域的热点。
2 ORR电催化与核壳结构催化剂
二十世纪九十年代,为深入理解电极材料与其表面小分子吸附能的关系,以Norskov为首4,5的理论电化学和物理学家基于计算化学成果提出了d带理论。这一简要却有效的理论在过去几十年里成功地从分子层面上解释了众多异相催化现象并指导了高效催化剂的结构设计,其中便包括了对于PEMFCs技术至关重要的阴极ORR。如前文所述,高Pt担量是制约PEMFCs规模化应用的主要障碍,而跨越这一障碍需要开发出更高活性的ORR催化剂。基于此d带理论,限制标准Pt/C催化剂表面ORR速率的关键因素在于其过强的含氧中间体吸附6-9。若能通过某种方式将纯Pt的d带中心降低,则能有效地降低其与含氧中间体的吸附能,进而提高ORR的催化活性10。基于这一理论指导,许多高性能低Pt量的电催化剂被设计和开发出来,而Pt基核壳结构催化剂便是其中最具代表性的一种。典型的核壳结构包括:单Pt原子层核壳结构11-15、表层富集核壳结构16-19、顺序还原与外延生长核壳结构20-22、脱合金核壳结构催化剂23-25和纳米多孔核壳结构催化剂26-28和化学有序核壳结构催化剂等。上述结构的一个共同特征是:外壳富Pt、内核富廉价过渡金属(例如Fe29-31、Co32-37、Ni38-42、Cu43-48、Y49-52、Gd53,54等)。这些廉价过渡金属除通过替代内核Pt降低成本外,还可通过合金作用或晶格应力使表层Pt原子的d带中心向费米能级方向移动,改善催化材料表面与含氧中间体的结合力,进而提高对ORR的催化活性。
除高ORR活性外,PEMFCs的阴极电催化剂还要求具备良好的稳定性和长久的寿命。目前为止,兼具活性和稳定性的催化材料设计仍是一个巨大的挑战,这主要是由于廉价过渡金属在PEMFCs阴极强酸性高电压的运行环境中易被侵蚀。变为离子形态的廉价过渡金属极易从阴极催化层扩散至质子交换膜或阳极催化层中,其在H2或低电压下被重新还原至金属价态并逐渐聚集为纳米簇或纳米颗粒,这对质子交换膜的离子传导率和机械性能将造成不可逆的损失,同时还会引起阳极催化剂的失活。例如,早期的许多研究都基于PtCu系列合金催化剂,因为此类合金催化剂在运行早期表现出极高的ORR催化性能。然而,在催化剂的稳定性研究中,人们发现Cu极易被脱去并使阳极催化剂快速失活55,最终导致PEMFCs的放电性能急剧降低。因此,高活性和高稳定性对于ORR电催化剂缺一不可。美国能源部56对车用PEMFCs阴极电催化剂提出了具体的技术目标,其中贵金属的比质量活性需达到0.44 A·mg-1以上,稳定性为:经过30000圈循环扫描后比质量活性的损失小于30%。本文将从催化剂的成本、活性和稳定性三个角度出发,综述几种典型的Pt基核壳结构电催化剂应用于PEMFCs阴极ORR的研究进展。
3 单Pt原子层催化剂
单Pt原子层概念最早由Adzic及其合作者15,57,58提出,旨在最大限度地提高贵金属Pt在电催化剂中的利用率及其对ORR的电催化活性。该结构催化剂具有极为精细的核壳结构,先后通过欠电位沉积和迦瓦尼置换的方式将单Pt原子层沉积至其他单一或合金基底的表面。为优化异相沉积效果,Adzic等59-61提出先将单原子层的Cu沉积到另一种氧化电势高于Cu还原电势的金属上,再通过迦瓦尼置换的方式将单原子层的Cu置换成Pt。众多理论和实验工作表明,该核壳结构催化剂具有极高的电化学活性面积,充分体现了单Pt原子层的结构优势,可大幅度节省Pt的用量。同时,单Pt原子层催化剂的基底物质对其整体催化性能有至关重要的影响,该影响主要来源于两个方面,(1)几何作用:由于原子半径的差异,基地物质会使单原子Pt层发生晶格收缩或扩张;(2)配位作用:近表层基底物质对Pt的直接合金化14。此二者皆能达到优化Pt层d带中心位置和氧结合能的目的,从而提高ORR的动力学速率。为深入研究基底对Pt原子层催化性能的影响,Zhang等62将单Pt原子层分别沉积至不同的单晶表面,理论和实验结果揭示了各表面的ORR活性与其d带中心位置呈火山型曲线关系(如图1所示)。其中,活性最高的催化剂为Pd(111)晶面上的单Pt原子层。Shao等63则发现以八面体Pd作为基底的单Pt原子层催化剂比以立方体Pd作为基底时的ORR催化活性高出3.5倍,该工作强调了基底物质高比例(111)晶面对Pt层催化活性的重要性。
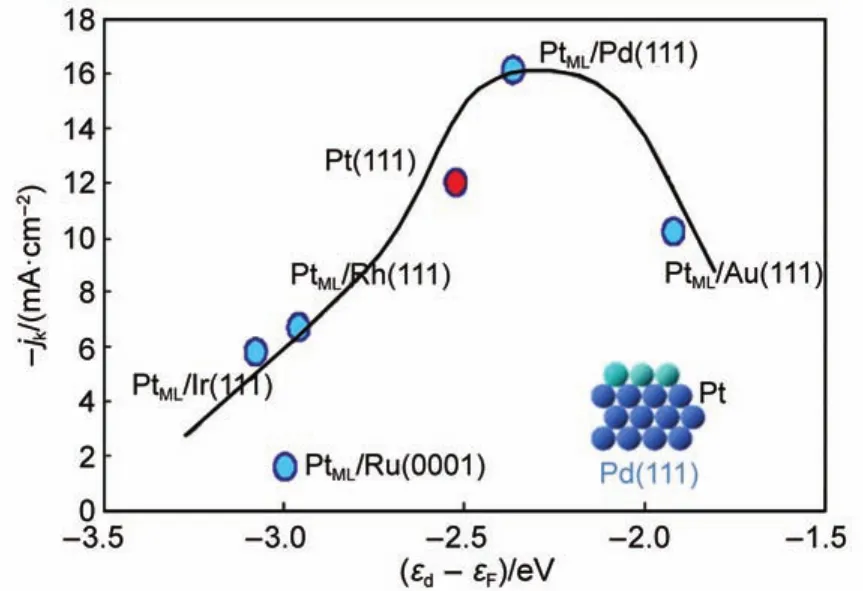
图1 不同电子结构单晶基底对单Pt原子层ORR催化活性的火山型曲线62Fig.1 Volcano plots:ORR activities of platinum monolayers on different single-crystal surfaces as functions of calculated d-band center62
由于异相沉积对基底物质的平整度要求较高,因此将单Pt原子层的策略移植到更实用的纳米催化剂时遇到了极大的问题。这是因为作为基底的纳米颗粒表面一般具有较多的缺陷和低配位原子,这些位点不仅妨碍异相沉积的完整性,还容易吸附含氧中间体并在催化剂的使用过程中优先流失,从而降低稳定性64。为解决这个问题,Cai等65通过Br预处理的方式移除Pd纳米颗粒表面的低配位原子,并以此为基底得到了光滑平整的单Pt原子层,该催化剂的(111)晶面比例得到了显著升高,其ORR催化活性比未经Br预处理的样品高出25%-50%。Gong等66则通过合成暴露晶面皆为(111)的四面体Pd纳米颗粒作为基底,得到了具有光滑Pt层的四面体核壳结构催化剂。该催化剂具有较高的活性,旋转圆盘电极得到的ORR极化曲线显示其半波电位相对商业Pt/C催化剂正移了20 mV。与纳米颗粒相比,纳米线/管具有更少的低配位原子数和更平整的晶面,因此有利于制备单Pt原子层催化剂。Koenigsmann等67以Pd纳米线(Pd-NW)为基底,成功制备了单Pt原子层的PtML/ Pd-NW催化剂,该催化剂在E=0.9 V(vs RHE(reversible hydrogen electrode))时的比质量活性高达1.83 A·mg-1,远超商业Pt/C催化剂和美国能源部所设定的2017年技术目标56。Shao等68发现在基底Pd表面引入Au作为中间层能进一步提高单Pt原子层的ORR活性。近日,华南理工大学廖世军课题组69首次采用TiNi合金的氮化物为基底和载体,以相同的技术制备得到了数个Pt原子层的核壳结构催化剂,其质量和面积比活性分别较商业Pt/C催化剂提高了4倍和2倍,经过10000圈循环扫描后只有极少量的性能损失。
以往多数单Pt原子层催化剂的制备都是在旋转圆盘电极上进行的,以致于只能得到微克级的催化剂样品。为推动该特殊结构催化剂的实用化进程,Zhou等70设计了特殊装置,以镀RuO2的金属钛板为工作电极,以镀Pt板为对电极,在实验室阶段批量制备了单Pt原子层的Pd2Co@Pt/C电催化剂。用此法制得的催化剂与用旋转圆盘电极合成的样品具有相似的电催化活性,证实了催化剂的物理和电子结构由模型和微量样品向批量制备样品的成功转换。然而由于大批量制备时对反应条件(主要包括电解液中Pt前驱体的浓度和扩散速率)的控制难度更高,很难保证所得样品在结构上的均一性。因此,寿命加速实验结果表明所得Pd2Co@Pt/C电催化剂的稳定性还有待提高。向催化剂中掺入极微量的Au则能大幅度提高催化剂的稳定性和寿命71。Sasaki等72在Pd9Au核上沉积了单Pt原子层制得了Pd9Au@Pt/C催化剂,其在0.6-1.0 V下循环扫描20000圈后,其Pt的比质量活性只损失了30%;当在0.6-1.4 V下循环扫描20000圈后,其Pt的比质量活性仍保留了30%,且核壳结构没有遭到破坏。
目前,单Pt原子层ORR催化剂的大批量制备技术日趋成熟,诸多不同元素配比的催化剂种类都展现出十分优越的活性和稳定性。与传统Pt/C催化剂(PEMFCs中的Pt载量一般为400 μg·cm-2)相比,单Pt原子层催化剂的Pt用量只需要40-80 μg· cm-2的Pt和60-100 μg·cm-2的Pd,极大地降低了PEMFCs阴极催化材料的成本73。然而,该结构催化剂还有进一步提高的空间,具体包括:(1)进一步降低基底中所用贵金属的含量(如Pd),选用更多的非贵金属元素;(2)改善基底表面数个原子层的结构,以进一步优化其对Pt原子层电子结构的作用;(3)设计及合成缺陷更少的基底物质,增加其(111)晶面的比例;(4)开发低成本的大规模制备方法;(5)优化膜电极的组装工艺以最大化单Pt原子层催化剂的性能。
4 脱合金催化剂
4.1脱合金简介
脱合金是通过化学或电化学方式选择性地将合金表面的活泼金属移除的过程,常用于制备多孔结构或核壳结构材料74,75。当活泼金属从表面移除后,裸露的惰性金属配位数会相应地降低,其迁移能力则相对变强,保护了活泼金属的进一步移除。因此,脱合金材料的最终形貌由活泼金属的溶解速率和惰性金属的迁移速率共同决定,当活泼金属的溶解速率大于惰性金属的迁移速率,则易得到多孔结构;反之,则易得到实心的核壳结构。由于材料本身的尺寸对表层惰性金属的迁移速率有较大影响,因此合金的原始尺寸也决定了脱合金材料的最终形貌。基于此原理,Gan等76在对PtNi3纳米颗粒进行脱合金研究时提出了一个临界尺寸(约为10 nm),当合金原始尺寸大于10 nm时,则易形成纳米多孔材料,小于10 nm则得到实心的核壳结构(如图2所示)。此外,脱合金过程中的气氛环境亦对最终形貌有重要影响。尽管Snyder等77发现纳米多孔结构PtNi催化剂的ORR催化活性要高于其对应的实心核壳结构,但前者远不如后者稳定,在测试过程中性能损失较快。本综述将分别介绍近年来通过脱合金法制备实心核壳结构催化剂和纳米多孔结构催化剂的研究进展。
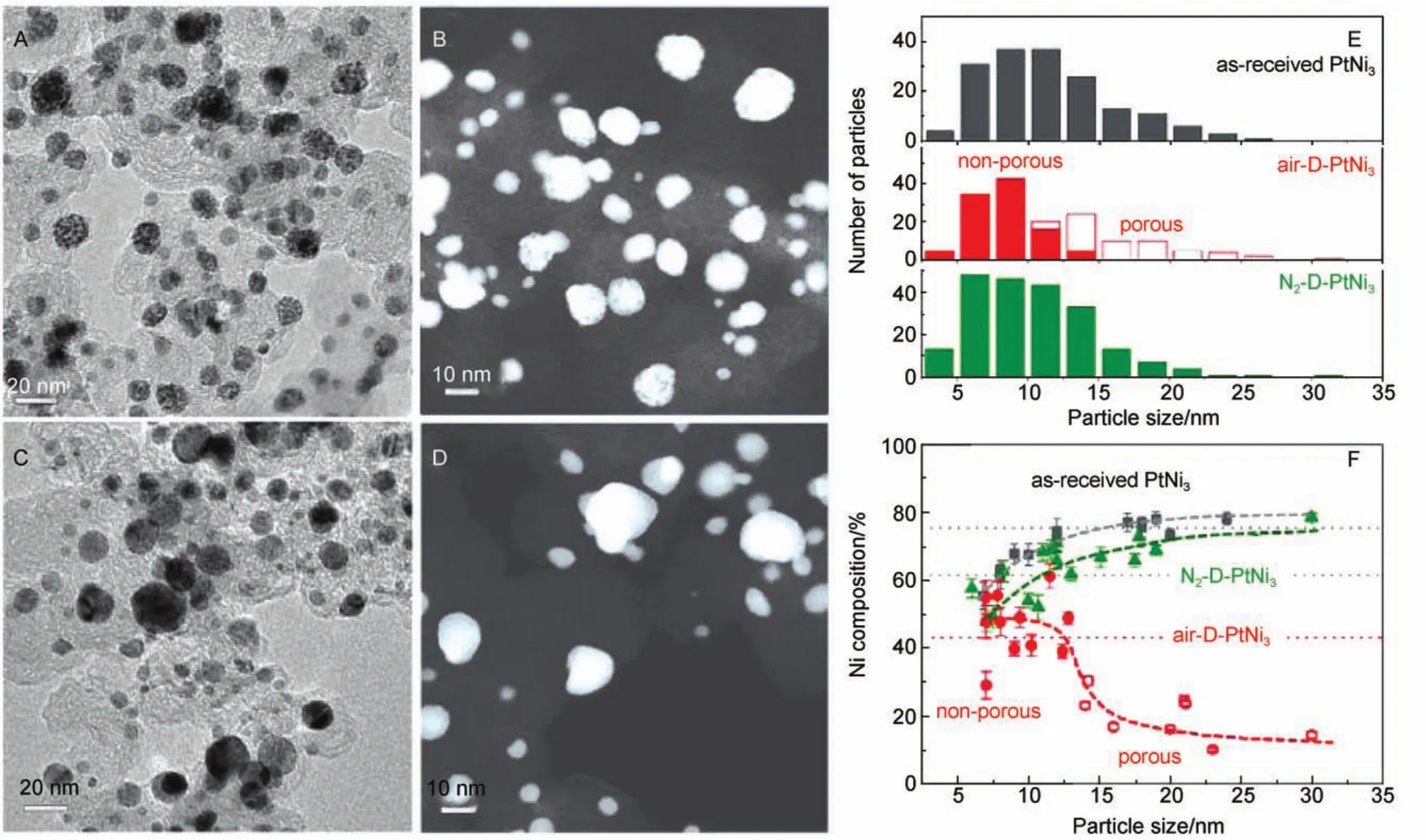
图2 空气环境脱合金PtNi3/C的(A)TEM和(B)HAADF-STEM图;氮气环境脱合金PtNi3/C的(C)TEM和(D)HAADF-STEM图;(E)各类PtNi3样品的粒径分布图及(F)催化剂粒径、成分和形貌之间的关系76Fig.2 (A)TEM and(B)HAADF-STEM image of air-D-PtNi3sample;(C)TEM and(D)HAADF-STEM images of N2-D-PtNi3sample;(E)particle size distributions of various PtNi3catalyst and (F)correlations between particle size,composition,and porosity76
4.2构效关系研究
采用脱合金法制备的实心核壳结构通常较纯Pt/C具有更高的ORR比催化活性,其原因被主要归结为壳层Pt原子的晶格收缩。Strasser等78以CuPt系列核壳结构催化剂为探针实验,采用电化学脱合金法得到了数个Pt原子层厚(0.6 nm)的壳,他们发现壳层Pt上的晶格间距略小于纯Pt,并认为这是由富Cu的内核所造成的晶格收缩现象。由于内核的Cu与最外层的Pt原子相隔数个原子层,所以在此体系中,Cu对Pt的配位作用(合金效应)可忽略不及。早期,该领域的研究多为利用电化学脱合金法(旋转圆盘电极或膜电极中)制备PtCu系列的ORR电催化剂,直到发现Cu在燃料电池运行中极易流失,电池性能损失较快79,才驱使人们转而研究稳定性更好的PtNi系列。因此,我们将主要介绍近年来Pt-Ni脱合金催化剂的研究进展。
4.3Pt-Ni脱合金核壳催化剂
过去十年里,纳米材料合成技术的快速发展使人们在Pt基纳米材料的可控制备方面(粒径、成分和形貌等)有了长足的发展,为PtNi脱合金催化剂的研究提供了基础80,81。Gan等82以Pt(acac)2和Ni(acac)2为前驱体,在油胺和油酸体系中合成了不同成分的PtNi、PtNi3和PtNi5纳米颗粒,将其负载到碳载体后得到了相应的催化剂。随后通过电化学脱合金方式(0.06-1.0 V扫描200圈)研究了原始合金成分对脱合金催化剂ORR活性的影响。结果发现PtNi3对应的脱合金催化剂展现出最高的ORR活性,其质量和面积比活性分别较商业Pt/C催化剂高出5和10倍。其活性顺序与催化剂中残留Ni的含量呈正比,进一步证明脱合金PtNi催化剂中内核Ni对ORR活性的重要影响,与Wang等83的结论一致。然而,在Wang的报道中,最高的Ni含量和活性为Pt1Ni1/C的脱合金样品而非Pt1Ni3/C,该结果说明催化剂的制备、预处理及脱合金方式对最终的ORR活性亦有重要影响。通过对样品的精细表征,人们进而发现与脱合金催化剂ORR活性直接关联的是其近表层的元素分布(包括Pt层厚度与Ni的近表层含量),而非整体的Ni含量。Wang等84在随后的报道中亦强调了脱合金Pt层厚度对其ORR催化性能的重要性:脱合金核壳催化剂中Pt壳层越厚,内核Ni对最外层Pt的晶格影响越小,其活性便越低。而Strasser等85则更强调近表层Ni (离表面10个原子层左右)含量对其活性的重要影响。在此,我们认为无法用单一的结构特征来解释合金原始比例对其脱合金样品ORR活性的影响规律,其本质还有待进一步的系统研究。
除PtNi的原始比例外,脱合金方式和操作条件对最终催化剂的结构、活性和稳定性也有至关重要的影响86。由于电化学脱合金需要在旋转圆盘电极或膜电极上进行,其制备量通常是毫克或微克级,显然无法满足燃料电池电极组装的要求,于是人们开始采用易批量合成的化学脱合金方式(强酸刻蚀)替代电化学脱合金。Han等87分别在热的稀硫酸和热的稀硝酸溶液中对PtNi3/C催化剂进行长达24 h的化学脱合金,他们发现硫酸处理得到的脱合金催化剂具有更厚的Pt壳和更高的Ni含量,其单电池的放电性能和稳定性亦更好(图3)。事实上,该脱合金催化剂亦保持着目前PEMFCs单电池性能测试的最高值,其活性和稳定性皆超越美国能源部所设定的单电池技术目标。同样,他们也发现当合金颗粒足够大时容易得到纳米多孔结构。本课题组通过化学脱合金方式制备了一系列高性能的催化剂88,同时也提出了批量化制备脱合金催化剂的工艺路线,在实验室阶段已实现了克级以上的合成,且重复性良好89。目前正致力于最大限度地提高PtxNiy系列脱合金催化剂中的Ni含量,从而得到更高活性的电催化剂。
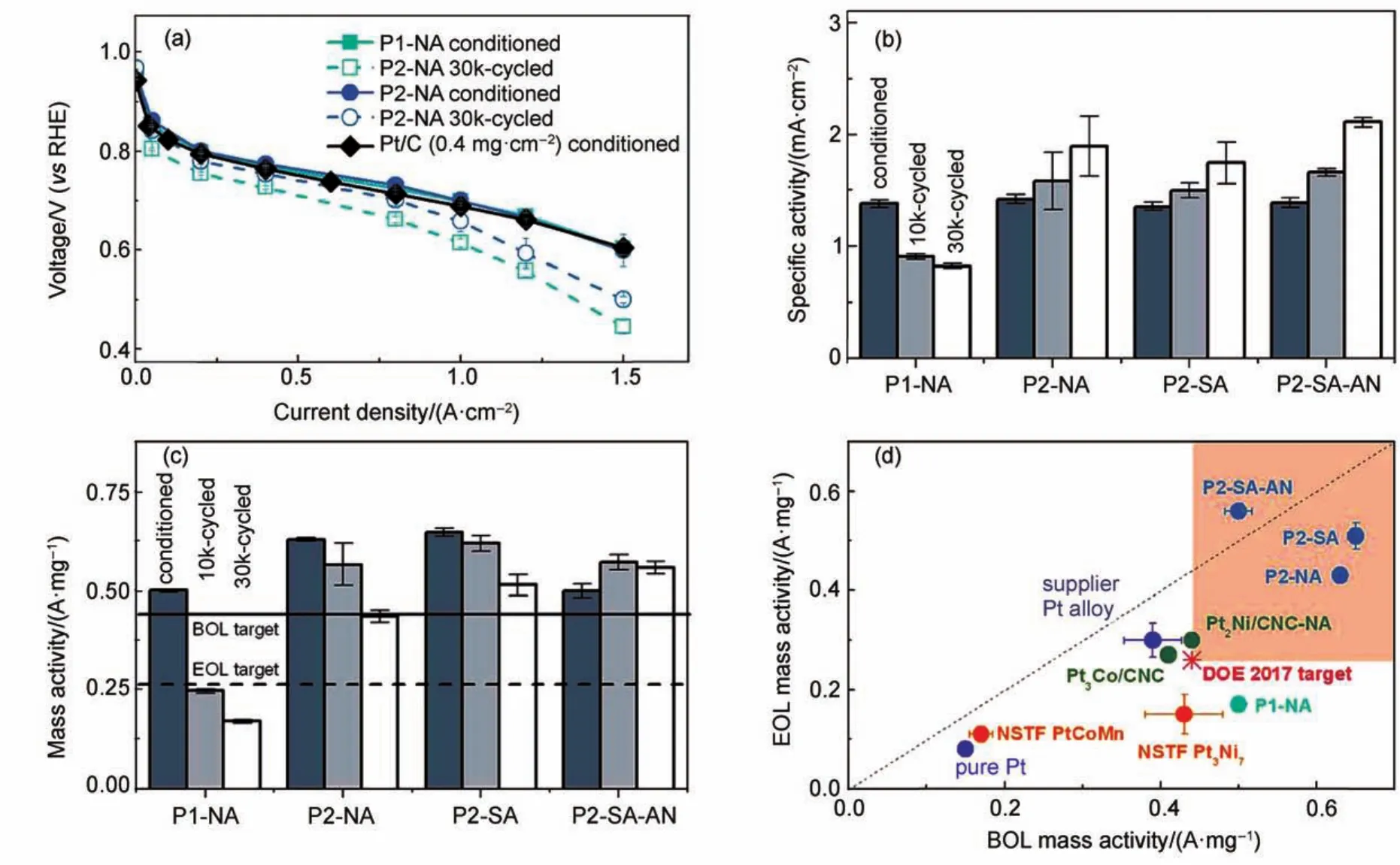
图3 (a)各阴极催化剂的单电池极化曲线及其(b)比面积活性和(c)比质量活性的对比柱状图;(d)膜电极上各催化剂稳定性测试前后的比质量活性对比87Fig.3 (a)Single cell polarization curves with P1-NAand P2-NAcathodes,(b)specific area activities and(c)specific mass activities of various dealloyed catalysts;(d)end-of-life(EOL)vs beginning-of-life(BOL)mass activity for various catalysts on membrane electrode assemblies(MEAs)87
4.4纳米多孔结构催化剂
相比于实心的核壳结构材料,纳米多孔催化剂有其特有的优势,如中空结构能提供更多的反应位点、纳米限域效应能提高其催化性能等90。2014年,Chen等91通过原子级的实验手段制备了一种被成为纳米框架催化剂的多孔结构。他们首先由化学还原法制得粒径约为20 nm的PtNi3多面体纳米颗粒,随后将其置于空气环境中的正己烷或氯仿中进行为期两周的缓慢脱合金过程,随后将其与碳载体结合并在400°C热处理得到更为光滑的Pt表面,最后对其进行电催化活性测试。Pt3Ni纳米框架催化剂的结构演变过程如图4所示。最初的PtNi3多面体为以(111)晶面为主的面心立方单晶结构,这主要是为了模仿由Markovic等16提出的具有超高活性(18 mA·cm-2)的Pt3Ni(111)晶面。最终的Pt3Ni框架结构由24条约为2 nm长的棱组成,每条棱皆类似于由脱合金制得的实心核壳结构。该结构催化剂的ORR质量和面积比活性较标准Pt/C分别提高了22和16倍,当向框架结构中填充有利于反应物O2传导的离子液体后,其催化活性得到了进一步提高。Becknell等92近日对Pt3Ni框架催化剂在实际工作中进行了原位的X射线光谱研究,他们指出该框架催化剂的活性和稳定性依赖于棱上Pt面的完整性。Wu等93则在PtNi纳米框架催化剂上引入了不同量的Au纳米簇。目前,纳米框架催化剂应用于实际PEMFCs中的研究工作正在进行中。
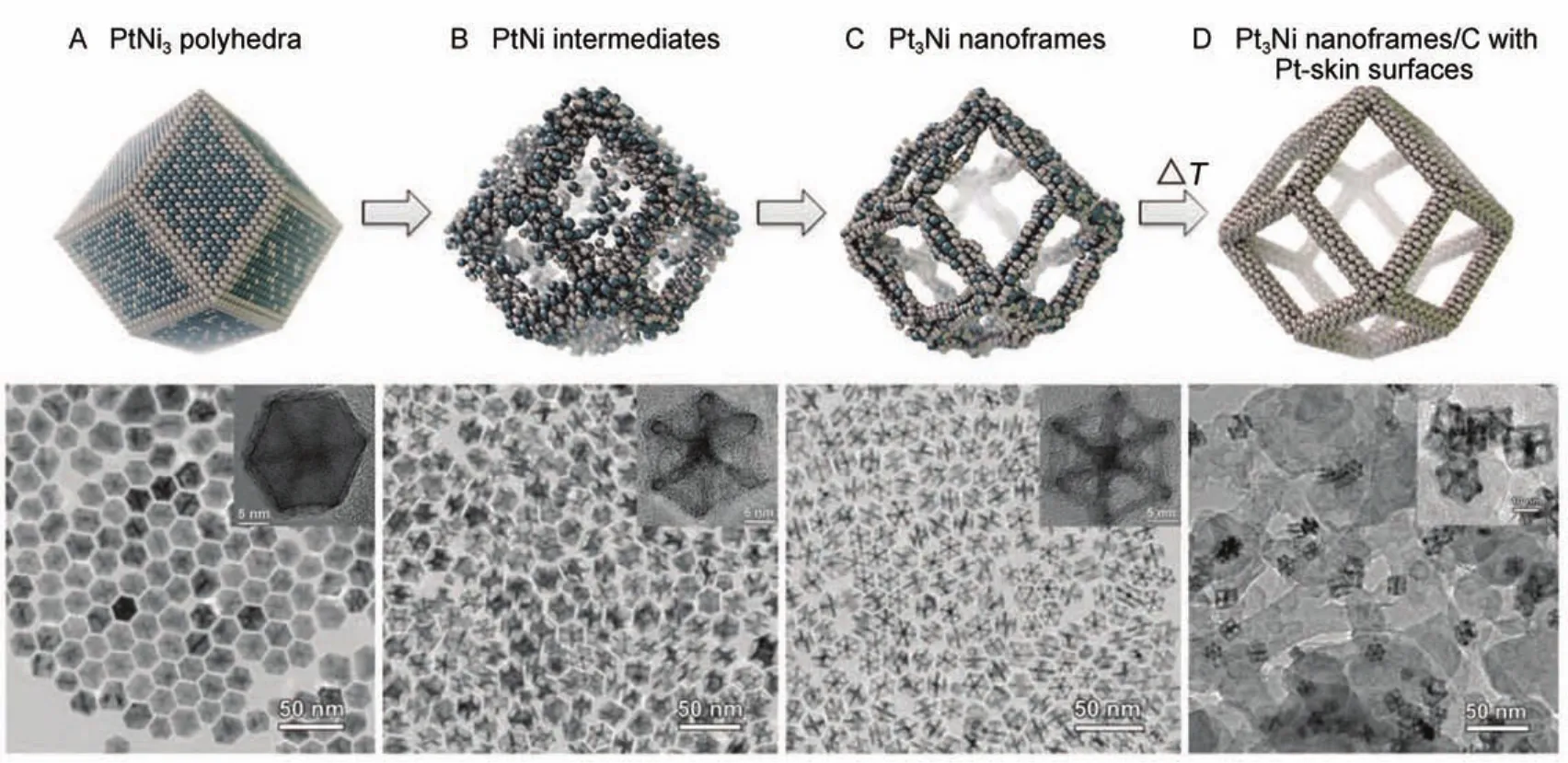
图4 纳米框架结构的演变示意图及TEM图片91Fig.4 Schematic diagrams and TEM images of the nano structures91
5 顺序还原与外延生长
顺序还原是制备核壳结构纳米材料最常用的方式之一。对于Pt基核壳结构,其采用适当的还原剂,在水相或有机相中,将Pt的前驱体盐还原沉积至已分散在反应体系中的纳米基底物表面。前人已研究了不同类型的基底物、溶剂、保护剂和还原剂等合成条件对形成核壳结构的影响94-98。Liu等99在乙二醇体系中,以聚乙烯吡咯烷酮(PVP)作为保护剂、数纳米大的Pd作为核,制得了均匀且较薄Pt层的Pd@Pt核壳结构催化剂,其壳层Pt的厚度可通过改变前驱体Pt盐的加入量来调节。然而,由于PVP难以被完全除掉,该催化剂没有表现出优异的ORR催化性能。大连化学物理研究所邵志刚研究员及其合作者100-102采用抗坏血酸作为还原剂、PEO106PPO70PEO106作为保护剂合成出不同Pd/Pt比例的Pd@Pt/C核壳结构催化剂。在初始的旋转圆盘电极测试中,该催化剂的活性依旧不高。但在单电池中循环40000圈后,其ORR活性得到了急剧升高,活性达到了Pt/C的4.5倍。该课题组还发现,由于内核的Pd会在扫描中缓慢流失,所合成的Pd@Pt催化剂经过电化学扫描后逐渐由实心的核壳结构变成空心的纳米笼结构,且后者的催化活性高于前者。Alia等103以PVP作为Pt的还原剂,在Pd纳米管表面能够沉积了数个Pt原子层,最薄Pt原子层的核壳结构具有最高的ORR催化活性。
Xia及其合作者104-107在采用顺序还原法制备Pd@Pt核壳结构材料的研究中提出了外延生长的概念。通过精确的实验控制,数个光滑的Pt原子层被包覆至Pd纳米立方体或八面体的表面,同样得到了较高的ORR比质量催化活性。为进一步降低贵金属用量,该课题组108将Pd@Pt浸于FeCl3/HCl溶液中将内核的Pd缓慢地腐蚀移除且不破坏Pt壳的完整性,得到了空心的立方纳米笼催化剂,遗憾的是其效果并不理想。Sun及其合作者42,109-111采用外延生长策略在Pd核或Au核表面制备了Pt基合金的壳层。首先,通过多元醇法制得了Pd、Au甚至Ni纳米颗粒,再将其充分分散于Pt(acac)2的溶液中。随后向上述混合液中加入Fe(CO)5,并进一步升温至200°C以分解铁盐。壳层合金含量(即Pt与Fe的比值)可通过前驱体的加入量进行调节。合金壳层的厚度亦通过前驱体的加入量进行调节,此工作实现了1-3 nm的调控。图5展示了Au@Pt3Fe纳米颗粒(其中Au为7 nm,Pt3Fe壳为1.5 nm)的合成示意图、透射电镜(TEM)及X射线衍射(XRD)。该催化剂的ORR比质量活性和比面积活性分别较标准Pt/C催化剂提高了5倍和3倍。除具有高活性外,Au@Pt3Fe/C催化剂还表现出极为优良的稳定性。经过60000圈0.6-1.1 V循环扫描后,Au@Pt3Fe/C样品的比质量活性和比面积活性分别是扫描后Pt/C催化剂的17倍和7倍。作者将稳定性提高的原因归结于Au核通过提高壳层金属的氧化电位而降低了流失速率。
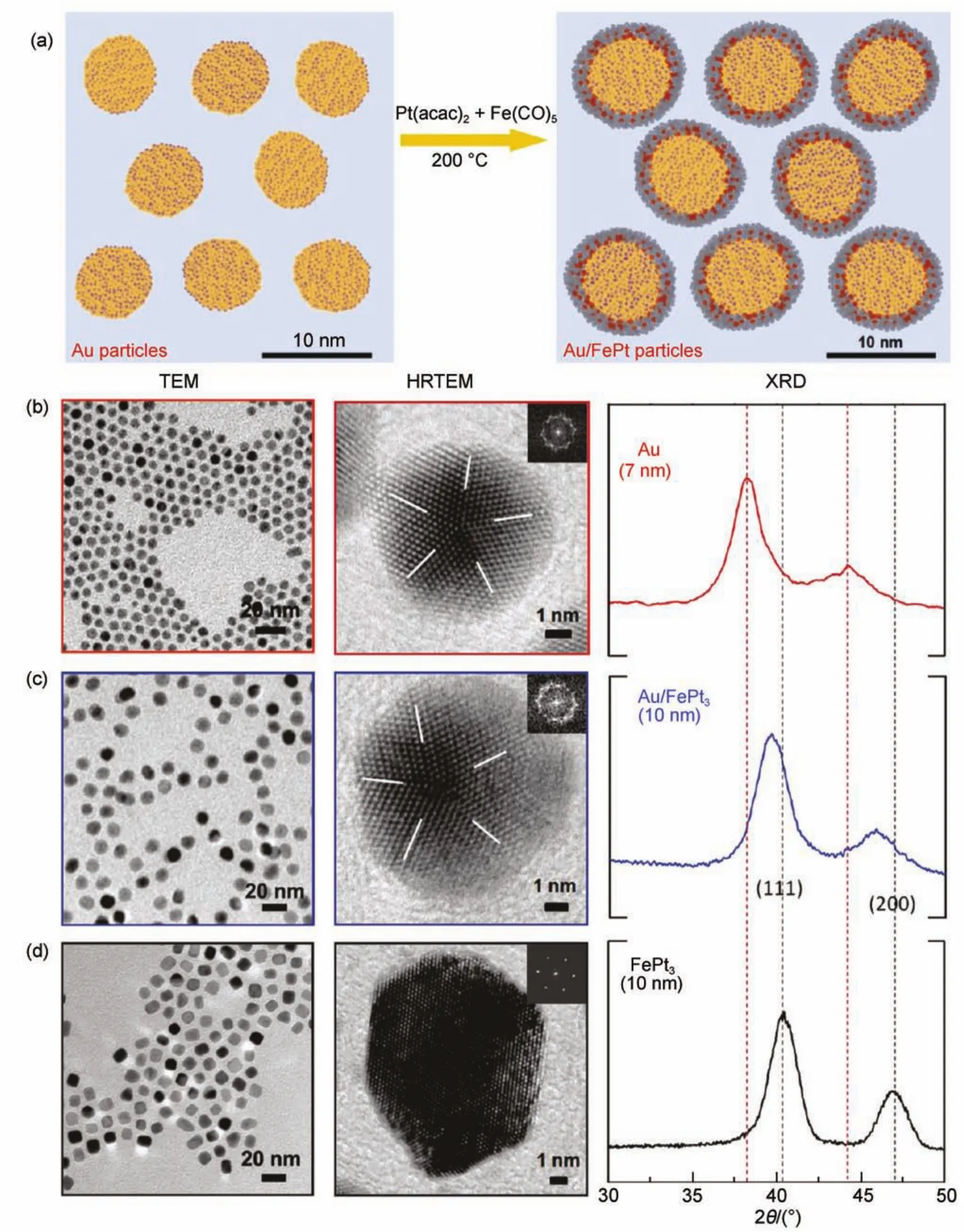
图5 (a)Au/FePt3核壳结构纳米颗粒的制备示意图;(b)7 nm的纯Au、(c)Au/FePt3和(d)10 nm的FePt3纳米颗粒的TEM、HRTEM和XRD表征109Fig.5 (a)Schematic illustration of the synthesis ofAu/FePt3NPs;TEM,HRTEM,and XRD characterizations of (b)7 nm pureAu,(c)Au/FePt3NPs,and(d)10 nm FePt3109
6 化学有序核壳结构催化剂
与化学无序的Pt基合金相比,化学有序的Pt基合金(或称为金属间化合物)具有高度合金化、均匀元素分布、特定电子结构、高催化活性和耐久性的优点112-114。为同时提高燃料电池阴极ORR催化剂中贵金属Pt的利用率及其催化活性、耐久性,人们研究了以金属间化合物为核、Pt层为壳的核壳结构化学有序Pt基催化剂115-117。
化学有序化合金的形成通常需要长时间高温热处理以克服固相里面的缓慢固-固相扩散速率,使得不同原子重新排布形成元素分布均匀的晶体结构。Wang等118首先采用浸渍法制备了Pt3Co/C合金催化剂,然后置于H2/N2混合气氛下,在400和700°C进行热处理分别得到化学无序的Pt3Co/C-400合金催化剂和一种以化学有序Pt3Co为核、2-3个Pt原子层为壳的化学有序核壳结构Pt3Co/C-700催化剂。电催化性能测试表明(如图6所示):两种PtCo合金催化剂的ORR活性明显高于商业Pt/C催化剂;而化学有序Pt3Co/C-700催化剂的比质量活性和比面积活性分别是化学无序Pt3Co/C-400催化剂的3.3倍和3.5倍。此外,较化学无序合金,化学有序核壳结构展示出更优良的稳定性,经5000圈循环扫描后,Pt3Co/C-700催化剂的ORR半波电位仅降低了约10 mV,电化学活性面积也只减少约20%,扫描后其结构没有发生明显变化。Zhang等119先在油胺和油酸体系中,采用Pt(acac)2和Fe(CO)5为前驱体,制得化学无序的fcc-FePt纳米颗粒及相应的碳载催化剂(FePt/C)。随后在H2/Ar的气氛中对FePt/C进行高温热处理,将无序fcc-FePt结构转变为有序fct-FePt结构,并通过脱合金方式得到壳厚约为0.6 nm的化学有序核壳结构催化剂。电化学性能测试表明,有序fct-FePt/C催化剂的比面积活性高达2.1 mA·cm-2,远高于商业Pt/ C和无序的fcc-FePt/C催化剂。

图6 (a)室温下各催化剂在0.1 mol·L-1HClO4中的ORR极化曲线;(b)Pt3Co/C-700在不同电势下的Koutecky-Levich曲线(插图为不同转速下的极化曲线);(c)各催化剂的比质量活性和(d)比面积活性比较118Fig.6 (a)ORR polarization curves for various samples in 0.1 mol·L-1HClO4;(b)Koutecky-Levich plots for Pt3Co/C-700(inset shows the corresponding polarization curves at different rotating speeds);comparison of (c)specific mass activity and(d)specific area activity for various catalysts118
为进一步提高Pt壳的催化活性,Wang等120结合密度泛函理论计算和实验研究结果提出在化学有序的AuCu表面沉积1-2个Pt原子层能有效改善氧结合能的强度,从而具有优良的ORR催化活性。他们首先在高沸点溶剂中直接合成了化学有序的AuCu纳米颗粒作为基底及相应的AuCu/C催化剂,然后将AuCu/C分散至K2PtCl4水溶液,使AuCu表面的Cu与Pt进行可控的置换取代,即得目标核壳结构。性能测试表明,该结构催化剂的ORR比质量活性较商业Pt/C提高了2.3倍。Bele等121提出了一次性可制备20g化学有序核壳结构催化剂的合成方法,进一步推动了该概念催化剂的批量应用。他们先通过溶胶凝胶法将Cu纳米颗粒镶入碳载体中,随后通过贾瓦尼置换得到CuPt无序合金,最后在还原气氛中得到目标产物。该催化剂的比活性高达2.45 mA·cm-2。
7 结论与展望
核壳结构催化剂不仅能提高贵金属Pt的利用率,还能通过配位或张力作用改善Pt壳的电子结构,从而提升催化活性和稳定性,是用于降低PEMFCs阴极电催化剂成本的有效策略。本文综述了几种典型的核壳结构在催化燃料电池阴极ORR上的应用研究,具体包括:单Pt原子层核壳结构、顺序还原与外延生长核壳结构、脱合金核壳结构催化剂、纳米多孔结构催化剂和化学有序核壳结构催化剂。基于目前的研究进展,我们认为将来核壳催化剂的研究应注重以下几个方面:
(1)优选更为廉价且稳定的“核”元素。常见的核壳结构催化剂中“核”的化学稳定性不够高,尽管有外层Pt壳的保护,在电催化过程中经常会流失。因此,优选廉价且稳定的“核”对核壳结构催化剂的发展至关重要。近日,Hunt等122通过可控自组装方式制备了以极其稳定的金属碳化物为核,数个Pt原子层为壳的电催化剂,在催化甲醇氧化和氢气氧化反应中表现出较好的活性和极高的稳定性。该工作可为寻找新型高效的“核”元素提供借鉴。
(2)电池放电性能测试。文献中大多数新结构催化剂的ORR催化活性测试皆只基于旋转圆盘电极装置,若要进一步推动高活性催化剂的实用化进程,则需将电催化剂组装至PEMFCs的电极中,测试其真实的放电性能。
(3)批量制备工艺研究。目前,许多核壳结构催化剂的活性和稳定性皆已满足美国能源部规定的技术目标,但将其放大制备时却遇到了诸如制备成本高、工艺复杂、结构不均一等问题。因此,一部分研究重心应放在催化剂批量制备工艺的开发和优化上。
(4)局部氧气扩散阻力。当PEMFCs阴极中的Pt担量降低至一定程度时,反应物氧气扩散至催化剂表面的阻力将急剧增加,这会使传质极化成为电池性能衰减的主要原因。
References
(3) Larminie,J.Fuel Cell Systems Explained;Science Press: Beijing,2003;pp 25-53;translated by Zhu,H.[Larminie,J.可再生能源开发技术.朱红,译.北京:科学出版社,2003: 25-53.]
(4) Hammer,B.;Norskov,J.K.Nature 1995,376,238. doi:10.1038/378376a0
(5) Mavrikakis,M.;Stoltze,P.;Norskov,J.K.Catal.Lett.2000, 64,101.doi:10.1023/A:1019028229377
(6) Nørskov,J.K.;Rossmeisl,J.;Logadottir,A.;Lindqvist,L.; Kitchin,J.R.;Bligaard,T.;Jónsson,H.J.Phys.Chem.B 2004, 108,17886.doi:10.1021/jp047349j
(7) Stamenkovic,V.;Mun,B.S.;Mayrhofer,K.J.;Ross,P.N.; Markovic,N.M.;Rossmeisl,J.;Greeley,J.;Norskov,J.K. Angew.Chem.Int.Ed.2006,45,2897.doi:10.1002/ anie.200504386
(8) Norskov,J.K.;Bligaard,T.;Rossmeisl,J.;Christensen,C.H. Nat.Chem.2009,1,37.doi:10.1038/NCHEM.121
(9) Viswanathan,V.;Hansen,H.A.;Rossmeisl,J.;Nørskov,J.K. ACS Catal.2012,2,1654.doi:10.1021/cs300227s
(10) Greeley,J.;Stephens,I.E.;Bondarenko,A.S.;Johansson,T. P.;Hansen,H.A.;Jaramillo,T.F.;Rossmeisl,J.;Chorkendorff, I.;Norskov,J.K.Nat.Chem.2009,1,552.doi:10.1038/ NCHEM.367
(11)Zhou,W.P.;Yang,X.;Vukmirovic,M.B.;Koel,B.E.;Jiao,J.; Peng,G.;Mavrikakis,M.;Adzic,R.R.J.Am.Chem.Soc. 2009,131,12755.doi:10.1021/ja9039746
(12) Wang,J.X.;Inada,H.;Wu,L.;Zhu,Y.;Choi,Y.;Liu,P.;Zhou, W.P.;Adzic,R.R.J.Am.Chem.Soc.2009,131,17298. doi:10.1021/ja9067645
(13) Shao,M.;Sasaki,K.;Marinkovic,N.;Zhang,L.;Adzic,R. Electrochem.Commun.2007,9,2848.doi:10.1016/j. elecom.2007.10.009
(14) Zhang,J.;Vukmirovic,M.B.;Sasaki,K.;Nilekar,A.U.; Mavrikakis,M.;Adzic,R.R.J.Am.Chem.Soc.2005,127, 12480.doi:10.1021/ja053695i
(15)Zhang,J.;Mo,Y.;Vukmirovic,M.B.;Klie,R.;Sasaki,K.; Adzic,R.R.J.Phys.Chem.B 2004,108,10955.doi:10.1021/ jp0379953
(16) Stamenkovic,V.R.;Fowler,B.;Mun,B.S.;Wang,G.;Ross,P. N.;Lucas,C.A.;Markovic,N.M.Science 2007,315,493. doi:10.1126/science.1135941
(17) Chen,S.;Ferreira,P.J.;Sheng,W.;Yabuuchi,N.;Allard,L.F.; Shao-Horn,Y.J.Am.Chem.Soc.2008,130,13818. doi:10.1021/ja802513y
(18) Stamenkovic,V.R.;Mun,B.S.;Arenz,M.;Mayrhofer,K.J.; Lucas,C.A.;Wang,G.;Ross,P.N.;Markovic,N.M.Nat. Mater.2007,6,241.doi:10.1038/nmat1840
(19)Stamenkovic,V.R.;Mun,B.S.;Mayrhofer,K.J.;Ross,P.N.; Markovic,N.M.J.Am.Chem.Soc.2006,128,8813. doi:10.1021/ja0600476
(20) Lim,B.;Jiang,M.;Camargo,P.H.;Cho,E.C.;Tao,J.;Lu,X.; Zhu,Y.;Xia,Y.N.Science 2009,324,1302.doi:10.1126/ science.1170377
(21) Harpeness,R.;Gedanken,A.Langmuir 2004,20,3431. doi:10.1021/la035978z
(22) Zhu,H.;Li,X.W.;Wang,F.H.Int.J.Hydrog.Energy 2011, 36,9151.doi:10.1016/j.ijhydene.2011.04.224
(23) Dutta,I.;Carpenter,M.K.;Balogh,M.P.;Ziegelbauer,J.M.; Moylan,T.E.;Atwan,M.H.;Irish,N.P.J.Phys.Chem.C Nanomater.Interfaces 2010,114,16309.doi:10.1021/ jp106042z
(24) Strasser,P.;Koh,S.;Greeley,J.Phys.Chem.Chem.Phys. 2008,10,3670.doi:10.1039/b803717e
(25) Mani,P.;Srivastava,R.;Strasser,P.J.Phys.Chem.C 2008, 112,2770.doi:10.1021/jp0776412
(26) Shviro,M.;Polani,S.;Zitoun,D.Nanoscale 2015,7,13521. doi:10.1039/C5NR03522H
(27) Dubau,L.;Asset,T.;Chattot,R.;Bonnaud,C.;Vanpeene,V.; Nelayah,J.;Maillard,F.ACS Catal.2015,5,5333. doi:10.1021/acscatal.5b01248
(28) Lee,C.L.;Huang,K.L.;Tsai,Y.L.;Chao,Y.J.Electrochem. Commun.2013,34,282.doi:10.1016/j.elecom.2013.07.020
(29)Chiwata,M.;Yano,H.;Ogawa,S.;Watanabe,M.;Iiyama,A.; Uchida,H.Electrochemistry 2016,84,133.doi:10.5796/ electrochemistry.84.133
(30)Wang,D.Y.;Chou,H.L.;Cheng,C.C.;Wu,Y.H.;Tsai,C. M.;Lin,H.Y.;Wang,Y.L.;Hwang,B.J.;Chen,C.C.Nano Energy 2015,11,631.doi:10.1016/j.nanoen.2014.11.040
(31)Wakabayashi,N.;Takeichi,M.;Uchida,H.;Watanabe,M. J.Phys.Chem.B 2005,109,5836.doi:10.1021/jp046204+
(32) Zhao,T.T.;Lin,R.;Zhang,L.;Cao,C.H.;Ma,J.X.Acta Phys.-Chim.Sin.2013,29,1745.[赵天天,林瑞,张路,曹春晖,马建新.物理化学学报,2013,29,1745.]doi:10.3866/ PKU.WHXB201305101
(33) Reyes-Rodríguez,J.L.;Godínez Salomón,F.;Leyva,M.A.; Solorza-Feria,O.Int.J.Hydrog.Energy 2013,38,12634. doi:10.1016/j.ijhydene.2012.12.031
(34)Matin,M.A.;Jang,J.H.;Kwon,Y.U.Int.J.Hydrog.Energy 2014,39,3710.doi:10.1016/j.ijhydene.2013.12.137
(35) Jia,Q.;Liang,W.;Bates,M.K.;Mani,P.;Lee,W.;Mukerjee, S.ACS Nano 2015,9,387.doi:10.1021/nn506721f
(36) Cao,C.H.;Lin,R.;Zhao,T.T.;Huang,Z.;Ma,J.X.Acta Phys.-Chim.Sin.2013,29,95.[曹春晖,林瑞,赵天天,黄真,马建新.物理化学学报,2013,29,95.]doi:10.3866/PKU. WHXB201209272
(37) Li,Z.;He,C.;Cai,M.;Kang,S.;Shen,P.K.Int.J.Hydrog. Energy 2012,37,14152.doi:10.1016/j.ijhydene.2012.07.100
(38) Morris,A.R.;Skoglund,M.D.;Holles,J.H.Appl.Catal.A: Gen.2015,489,98.doi:10.1016/j.apcata.2014.10.019
(39) Ramos-Sanchez,G.;Praserthdam,S.;Godinez-Salomon,F.; Barker,C.;Moerbe,M.;Calderon,H.A.;Lartundo,L.A.; Leyva,M.A.;Solorza-Feria,O.;Balbuena,P.B.Phys.Chem. Chem.Phys.2015,17,28286.doi:10.1039/c5cp00503e
(41) Chen,Y.;Shi,J.J.Fuel Cell Sci.Tech.2014,12,021005. doi:10.1115/1.4028149
(42) Zhang,S.;Hao,Y.;Su,D.;Doan-Nguyen,V.V.;Wu,Y.;Li,J.; Sun,S.;Murray,C.B.J.Am.Chem.Soc.2014,136,15921. doi:10.1021/ja5099066
(43) Dhavale,V.M.;Kurungot,S.ACS Catal.2015,5,1445. doi:10.1021/cs501571e
(44)Zhu,C.M.;Gao,A.;Wang,Y.;Liu,Y.Chem.Commun.2014, 50,13889.doi:10.1039/C4CC02391A
(45) Wang,D.;Yu,Y.;Zhu,J.;Liu,S.;Muller,D.A.;Abruna,H.D. Nano Lett.2015,15,1343.doi:10.1021/nl504597j
(46) Han,L.;Liu,H.;Cui,P.;Peng,Z.;Zhang,S.;Yang,J.Sci.Rep. 2014,4,6414.doi:10.1038/srep06414
(47) Xu,Z.;Zhang,H.;Liu,S.;Zhang,B.;Zhong,H.;Su,D.S.Int. J.Hydrog.Energy 2012,37,17978.doi:10.1016/j. ijhydene.2012.09.050
(48) Xu,C.;Liu,Y.;Wang,J.;Geng,H.;Qiu,H.ACS Appl.Mater. Interfaces 2011,3,4626.doi:10.1021/am201057t
(49) Johansson,T.P.;Ulrikkeholm,E.T.;Hernandez-Fernandez,P.; Escudero-Escribano,M.;Malacrida,P.;Stephens,I.E.; Chorkendorff,I.Phys.Chem.Chem.Phys.2014,16,13718. doi:10.1039/C4CP00319E
(50) Escudero-Escribano,M.;Malacrida,P.;Hansen,M.H.;Vej-Hansen,U.G.;Velazquez-Palenzuela,A.;Tripkovic,V.; Schiotz,J.;Rossmeisl,J.;Stephens,I.E.;Chorkendorff,I. Science 2016,352,73.doi:10.1126/science.aad8892
(51) Malacrida,P.;Casalongue,H.G.;Masini,F.;Kaya,S.; Hernandez-Fernandez,P.;Deiana,D.;Ogasawara,H.; Stephens,I.E.;Nilsson,A.;Chorkendorff,I.Phys.Chem.Chem.Phys.2015,17,28121.doi:10.1039/C5CP00283D
(52) Hernandez-Fernandez,P.;Masini,F.;McCarthy,D.N.;Strebel, C.E.;Friebel,D.;Deiana,D.;Malacrida,P.;Nierhoff,A.; Bodin,A.;Wise,A.M.;Nielsen,J.H.;Hansen,T.W.;Nilsson, A.;Stephens,I.E.;Chorkendorff,I.Nat.Chem.2014,6,732. doi:10.1038/NCHEM.2001
(53) Velázquez-Palenzuela,A.;Masini,F.;Pedersen,A.;Escudero-Escribano,M.;Deiana,D.;Malacrida,P.;Hansen,T.W.; Friebel,D.;Nilsson,A.;Stephens,I.E.L.;Chorkendorff,I.B. J.Catal.2015,328,297.doi:10.1016/j.jcat.2014.12.012
(54) Escudero-Escribano,M.;Verdaguer Casadevall,A.;Malacrida, P.;Gronbjerg,U.;Knudsen,B.P.;Jepsen,A.K.;Rossmeisl,J.; Stephens,I.E.;Chorkendorff,I.J.Am.Chem.Soc.2012,134, 16476.doi:10.1021/ja306348d
(55) Jia,Q.;Ramaker,D.E.;Ziegelbauer,J.M.;Ramaswamy,N.; Halder,A.;Mukerjee,S.J.Phys.Chem.C 2013,117,4585. doi:10.1021/jp311353u
(56) U.S.Department of Energy.Multi-year Research,Development and Demonstration Plan:Planned ProgramActivities for 2005-2015.2009.
(57)Adzic,R.R.;Zhang,J.;Sasaki,K.;Vukmirovic,M.B.;Shao, M.;Wang,J.X.;Nilekar,A.U.;Mavrikakis,M.;Valerio,J.A.; Uribe,F.Top.Catal.2007,46,249.doi:10.1007/s11244-007-9003-x
(58) Brankovic,S.R.;Wang,J.X.;Adžić,R.R.Electrochem.Solid-State Lett.2001,4,A217.doi:10.1149/1.1414943
(59) Zhang,Y.;Ma,C.;Zhu,Y.;Si,R.;Cai,Y.;Wang,J.X.;Adzic, R.R.Catal.Today 2013,202,50.doi:10.1016/j. cattod.2012.03.040
(60) Kuttiyiel,K.A.;Sasaki,K.;Choi,Y.;Su,D.;Liu,P.;Adzic,R. R.Energy Environ.Sci.2012,5,5297.doi:10.1039/ C1EE02067F
(61) Gong,K.;Su,D.;Adzic,R.R.J.Am.Chem.Soc.2010,132, 14364.doi:10.1021/ja1063873
(62)Zhang,J.;Vukmirovic,M.B.;Xu,Y.;Mavrikakis,M.;Adzic, R.R.Angew.Chem.Int.Ed.2005,44,2132.doi:10.1002/ anie.200462335
(63) Shao,M.H.;He,G.;Peles,A.;Odell,J.H.;Zeng,J.;Su,D.; Tao,J.;Yu,T.;Zhu,Y.;Xia,Y.N.Chem.Commun.2013,49, 9030.doi:10.1039/C3CC43276A
(64) Wang,J.X.;Ma,C.;Choi,Y.;Su,D.;Zhu,Y.;Liu,P.;Si,R.; Vukmirovic,M.B.;Zhang,Y.;Adzic,R.R.J.Am.Chem.Soc. 2011,133,13551.doi:10.1021/ja204518x
(65)Cai,Y.;Ma,C.;Zhu,Y.;Wang,J.X.;Adzic,R.R.Langmuir 2011,27,8540.doi:10.1021/la200753z
(66)Gong,K.;Vukmirovic,M.B.;Ma,C.;Zhu,Y.;Adzic,R.R. J.Electroanal.Chem.2011,662,213.doi:10.1016/j. jelechem.2011.07.008
(67) Koenigsmann,C.;Santulli,A.C.;Gong,K.;Vukmirovic,M. B.;Zhou,W.P.;Sutter,E.;Wong,S.S.;Adzic,R.R.J.Am. Chem.Soc.2011,133,9783.doi:10.1021/ja111130t
(68) Shao,M.H.;Peles,A.;Odell,J.J.Phys.Chem.C 2014,118, 18505.doi:10.1021/jp503296s
(69) Tian,X.L.;Luo,J.M.;Nan,H.X.;Zou,H.B.;Chen,R.;Shu, T.;Li,X.H.;Li,Y.W.;Song,H.Y.;Liao,S.J.;Adzic,R.R. J.Am.Chem.Soc.2016,138,1575.doi:10.1021/jacs.5b11364
(70) Zhou,W.P.;Sasaki,K.;Su,D.;Zhu,Y.;Wang,J.X.;Adzic,R. R.J.Phys.Chem.C 2010,114,8950.doi:10.1021/jp100283p
(71) Zhang,J.;Sasaki,K.;Sutter,E.;Adzic,R.R.Science 2007, 315,220.doi:10.1126/science.1134569
(72) Sasaki,K.;Naohara,H.;Choi,Y.;Cai,Y.;Chen,W.F.;Liu,P.; Adzic,R.R.Nat.Commun.2012,3,1115.doi:10.1038/ ncomms2124
(74) Erlebacher,J.;Aziz,M.J.;Karma,A.;Dimitrov,N.;Sieradzki, K.Nature 2001,410,450.doi:10.1038/35068529
(75) Wang,D.S.;Zhao,P.;Li,Y.D.Sci.Rep.2011,1,37. doi:10.1038/srep00037
(76) Gan,L.;Heggen,M.;O′Malley,R.;Theobald,B.;Strasser,P. Nano Lett.2013,13,1131.doi:10.1021/nl304488q
(77) Snyder,J.;McCue,I.;Livi,K.;Erlebacher,J.J.Am.Chem. Soc.2012,134,8633.doi:10.1021/ja3019498
(78) Strasser,P.;Koh,S.;Anniyev,T.;Greeley,J.;More,K.;Yu,C.; Liu,Z.;Kaya,S.;Nordlund,D.;Ogasawara,H.;Toney,M.F.; Nilsson,A.Nat.Chem.2010,2,454.doi:10.1038/nchem.623
(79) Hasché,F.;Oezaslan,M.;Strasser,P.ChemCatChem 2011,3, 1805.doi:10.1002/cctc.201100169
(80) Skrabalak,S.E.;Xia,Y.N.ACS Nano 2009,3,10. doi:10.1021/nn800875p
(82) Gan,L.;Heggen,M.;Rudi,S.;Strasser,P.Nano Lett.2012, 12,5423.doi:10.1021/nl302995z
(83) Wang,C.;Chi,M.;Wang,G.;van der Vliet,D.;Li,D.;More, K.;Wang,H.H.;Schlueter,J.A.;Markovic,N.M.; Stamenkovic,V.R.Adv.Funct.Mater.2011,21,147. doi:10.1002/adfm.201001138
(84) Wang,C.;Chi,M.;Li,D.;Strmcnik,D.;van der Vliet,D.; Wang,G.;Komanicky,V.;Chang,K.C.;Paulikas,A.P.; Tripkovic,D.;Pearson,J.;More,K.L.;Markovic,N.M.; Stamenkovic,V.R.J.Am.Chem.Soc.2011,133,14396. doi:10.1021/ja2047655
(85) Gan,L.;Cui,C.;Rudi,S.;Strasser,P.Top.Catal.2013,57, 236.doi:10.1007/s11244-013-0178-z
(86) Caldwell,K.M.;Ramaker,D.E.;Jia,Q.;Mukerjee,S.; Ziegelbauer,J.M.;Kukreja,R.S.;Kongkanand,A.J.Phys. Chem.C 2015,119,757.doi:10.1021/jp5098553
(87) Han,B.;Carlton,C.E.;Kongkanand,A.;Kukreja,R.S.; Theobald,B.R.;Gan,L.;O′Malley,R.;Strasser,P.;Wagner,F. T.;Shao-Horn,Y.Energy Environ.Sci.2015,8,258. doi:10.1039/c4ee02144d
(88)Zhu,H.;Luo,M.C.;Zhang,S.;Wei,L.L.;Wang,F.H.;Wang, Z.M.;Wei,Y.S.;Han,K.F.Int.J.Hydrog.Energy 2013,38, 3323.doi:10.1016/j.ijhydene.2012.12.127
(89)Luo,M.C.;Wei,L.L.;Wang,F.H.;Han,K.F.;Zhu,H. J.Power Sources 2014,270,34.doi:10.1016/j. jpowsour.2014.07.102
(90)Bae,J.H.;Han,J.H.;Chung,T.D.Phys.Chem.Chem. Phys.2012,14,448.doi:10.1039/c1cp22927c
(91) Chen,C.;Kang,Y.;Huo,Z.;Zhu,Z.;Huang,W.;Xin,H.L.; Snyder,J.D.;Li,D.;Herron,J.A.;Mavrikakis,M.;Chi,M.; More,K.L.;Li,Y.;Markovic,N.M.;Somorjai,G.A.;Yang, P.;Stamenkovic,V.R.Science 2014,343,1339.doi:10.1126/ science.1249061
(92) Becknell,N.;Kang,Y.;Chen,C.;Resasco,J.;Kornienko,N.; Guo,J.;Markovic,N.M.;Somorjai,G.A.;Stamenkovic,V. R.;Yang,P.J.Am.Chem.Soc.2015,137,15817.doi:10.1021/ jacs.5b09639
(93)Wu,Y.;Wang,D.;Zhou,G.;Yu,R.;Chen,C.;Li,Y.J.Am. Chem.Soc.2014,136,11594.doi:10.1021/ja5058532
(94) Jung,N.;Chung,Y.H.;Chung,D.Y.;Choi,K.H.;Park,H.Y.; Ryu,J.;Lee,S.Y.;Kim,M.;Sung,Y.E.;Yoo,S.J.Phys. Chem.Chem.Phys.2013,15,17079.doi:10.1039/c3cp52807c
(95)Du,B.;Zaluzhna,O.;Tong,Y.J.Phys.Chem.Chem.Phys. 2011,13,11568.doi:10.1039/c1cp20761j
(96)Atienza,D.O.;Allison,T.C.;Tong,Y.J.J.Phys.Chem.C 2012,116,26480.doi:10.1021/jp310313k
(97) Chen,Y.;Liang,Z.;Yang,F.;Liu,Y.;Chen,S.J.Phys.Chem. C 2011,115,24073.doi:10.1021/jp207828n
(98)Long,N.V.;Ohtaki,M.;Hien,T.D.;Randy,J.;Nogami,M. Electrochim.Acta 2011,56,9133.doi:10.1016/j. electacta.2011.07.090
(99) Liu,L.;Samjeske,G.;Nagamatsu,S.I.;Sekizawa,O.; Nagasawa,K.;Takao,S.;Imaizumi,Y.;Yamamoto,T.;Uruga, T.;Iwasawa,Y.J.Phys.Chem.C 2012,116,23453. doi:10.1021/jp308021a
(100) Zhang,G.;Shao,Z.G.;Lu,W.;Xie,F.;Qin,X.;Yi,B. Electrochim.Acta 2013,103,66.doi:10.1016/j. electacta.2013.04.045
(101) Zhang,G.;Shao,Z.G.;Lu,W.;Xiao,H.;Xie,F.;Qin,X.;Li, J.;Liu,F.;Yi,B.J.Phys.Chem.C 2013,117,13413. doi:10.1021/jp401375b
(102) Zhang,G.;Shao,Z.G.;Lu,W.;Xie,F.;Xiao,H.;Qin,X.;Yi, B.Appl.Catal.B:Environ.2013,132-133,183.doi:10.1016/ j.apcatb.2012.11.029
(103)Alia,S.M.;Jensen,K.O.;Pivovar,B.S.;Yan,Y.S.ACS Catal.2012,2,858.doi:10.1021/cs200682c
(104)Zhang,H.;Jin,M.;Wang,J.;Kim,M.J.;Yang,D.;Xia,Y. J.Am.Chem.Soc.2011,133,10422.doi:10.1021/ja204447k
(105)Zhang,H.;Jin,M.;Wang,J.;Li,W.;Camargo,P.H.;Kim,M. J.;Yang,D.;Xie,Z.;Xia,Y.J.Am.Chem.Soc.2011,133, 6078.doi:10.1021/ja201156s
(106) Jiang,M.;Lim,B.;Tao,J.;Camargo,P.H.C.;Ma,C.;Zhu,Y.; Xia,Y.Nanoscale 2010,2,2406.doi:10.1039/c0nr00324g
(107)Lim,B.;Wang,J.;Camargo,P.H.;Jiang,M.;Kim,M.J.;Xia, Y.Nano Lett.2008,8,2535.doi:10.1021/nl8016434
(108)Zhang,L.;Roling,L.T.;Wang,X.;Vara,M.;Chi,M.;Liu,J.; Choi,S.I.;Park,J.;Herron,J.A.;Xie,Z.;Mavrikakis,M.; Xia,Y.N.Science 2015,349,412.doi:10.1126/science. aab0801
(109) Wang,C.;van der Vliet,D.;More,K.L.;Zaluzec,N.J.;Peng, S.;Sun,S.;Daimon,H.;Wang,G.;Greeley,J.;Pearson,J.; Paulikas,A.P.;Karapetrov,G.;Strmcnik,D.;Markovic,N.M.; Stamenkovic,V.R.Nano Lett.2011,11,919.doi:10.1021/ nl102369k
(110)Mazumder,V.;Chi,M.;More,K.L.;Sun,S.H.J.Am.Chem. Soc.2010,132,7848.doi:10.1021/ja1024436
(111)Mazumder,V.;Chi,M.;More,K.L.;Sun,S.H.Angew.Chem. Int.Ed.2010,49,9368.doi:10.1002/anie.201003903
(112)Meku,E.;Du,C.;Sun,Y.;Du,L.;Wang,Y.;Yin,G. J.Electrochem.Soc.2016,163,F132.doi:10.1149/ 2.0031603jes
(113) Kumar,V.B.;Sanetuntikul,J.;Ganesan,P.;Porat,Z.E.; Shanmugam,S.;Gedanken,A.Electrochim.Acta 2016,190, 659.doi:10.1016/j.electacta.2015.12.193
(114) Cui,Z.;Li,L.;Manthiram,A.;Goodenough,J.B.J.Am. Chem.Soc.2015,137,7278.doi:10.1021/jacs.5b03865
(115) Cui,Z.;Chen,H.;Zhou,W.;Zhao,M.;DiSalvo,F.J.Chem. Mater.2015,27,7538.doi:10.1021/acs.chemmater.5b03912
(116)Chung,D.Y.;Jun,S.W.;Yoon,G.;Kwon,S.G.;Shin,D.Y.; Seo,P.;Yoo,J.M.;Shin,H.J.;Chung,Y.H.;Kim,H.;Mun,B. S.;Lee,K.;Lee N.S.;Yoo,S.J.;Lim,D.;Kang,K.;Sung,Y.; Hyeon,T.J.Am.Chem.Soc.2015,137,15478.doi:10.1021/ jacs.5b09653
(117)Arumugam,B.;Tamaki,T.;Yamaguchi,T.ACS Appl.Mater. Interfaces 2015,7,16311.doi:10.1021/acsami.5b03137
(118)Wang,D.L.;Xin,H.L.;Hovden,R.;Wang,H.;Yu,Y.;Muller, D.A.;DiSalvo,F.J.;Abruña,H.D.Nat.Mater.2012,12,81. doi:10.1038/nmat3458
(119) Zhang,S.;Zhang,X.;Jiang,G.;Zhu,H.;Guo,S.J.;Su,D.; Lu,G.;Sun,S.H.J.Am.Chem.Soc.2014,136,7734. doi:10.1021/ja5030172
(120)Wang,G.;Huang,B.;Xiao,L.;Ren,Z.;Chen,H.;Wang,D. L.;Abruna,H.D.;Lu,J.;Zhuang,L.J.Am.Chem.Soc.2014, 136,9643.doi:10.1021/ja503315s
(121) Bele,M.;Jovanovic,P.;Pavlisic,A.;Jozinovic,B.;Zorko,M.; Recnik,A.;Chernyshova,E.;Hocevar,S.;Hodnik,N.; Gaberscek,M.Chem.Commun.2014,50,13124.doi:10.1039/ C4CC05637J
(122)Hunt,S.T.;Milina,M.;Alba-Rubio,A.C.;Hendon C.H.; Dumesic,J.A.;Roman-Leshkov,Y.Science 2016,352,974. doi:10.1126/science.aad8471
Core-Shell Structured Electrocatalysts for the Cathodic Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells
ZHU Hong*LUO Ming-ChuanCAI Ye-ZhengSUN Zhao-Nan
(State Key Laboratory of Chemical Resource Engineering,School of Science,Beijing University of Chemical Technology, Beijing 100029,P.R.China)
Proton exchange membrane fuel cells(PEMFCs)are considered as ideal alternative power devices to traditional internal combustion engines for automobile applications because of their high electric power density, high energy conversion efficiency,and low environmental impact as well as low temperatures for start-up and operation.However,PEMFCs normally require a high loading of the expensive precious metal platinum(Pt) as the electrocatalytic material to maintain desirable energy output.Thus,the development of novel catalysts with lower Pt loading,enhanced activity,and improved durability is vital for the scalable commercialization of PEMFC technology.In this regard,core-shell structure has been demonstrated as an effective strategy to minimize the amount of Pt in PEMFCs because of the following two factors:(1)a core-shell architecture with a Pt-rich shell and M-rich(M represents an earth-abundant element)core can greatly improve the utilization of Pt;(2)the activity and stability of Pt on the surface can be greatly enhanced by strain(geometry)and electronic (alloying)effects caused by the M in the core.First,we briefly discuss the structure-performance relationship of typical core-shell structured electrocatalysts for the oxygen reduction reaction(ORR).Then,we review the development of Pt-based core-shell structured catalysts for the ORR.Finally,a perspective on this research topic is provided.
Proton exchange membrane fuel cell;Oxygen reduction reaction;Low-platinum catalyst; Core-shell structure;Electronic/geometric effect
May 9,2016;Revised:June 27,2016;Published online:June 29,2016.
.Email:zhuho128@126.com;Tel:+86-10-64444919.
O646
10.3866/PKU.WHXB201606293
The project was supported by the National Natural Science Foundation of China(21376022)and International S&T Cooperation Program of China (2013DFA51860).
国家自然科学基金(21376022)及国际合作项目(2013DFA51860)资助©Editorial office ofActa Physico-Chimica Sinica
(1) Gates,B.Science 2011,334,877.10.1126/ science.1216290
(2) Debe,M.K.Nature 2012,486,43.10.1038/nature11115
(40)An,W.;Liu,P.ACS Catal.2015,5,6328.10.1021/ acscatal.5b01656
(73) Adzic,R.R.Electrocatalysis-Us 2012,3,163.10.1007/ s12678-012-0112-3
(81) Peng,Z.;Yang,H.Nano Today 2009,4,143.10.1016/j. nantod.2008.10.010
