甘油催化氢解制备精细化学品的研究进展
2016-11-18朱善辉王建国樊卫斌
朱善辉王建国 樊卫斌
(中国科学院山西煤炭化学研究所,煤转化国家重点实验室,太原 030001)
甘油催化氢解制备精细化学品的研究进展
朱善辉*王建国 樊卫斌*
(中国科学院山西煤炭化学研究所,煤转化国家重点实验室,太原 030001)
随着生物柴油产业的快速发展,作为副产物的甘油大量过剩,因而有效利用甘油既能促进生物柴油产业的良性发展,又能节约大量石油资源。通过甘油催化氢解的方式来制备高附加值化学品丙二醇、乙二醇和丙醇等是甘油转化研究中最有潜在应用价值的路径之一,甘油氢解反应易于实现连续化生产,且目标产物附加值高、选择性高,因而具有良好的经济效益。本文首先简要介绍了甘油化学,深入探讨了甘油的氢解机理,然后重点综述了甘油氢解制备1,2-丙二醇、1,3-丙二醇、乙二醇和丙醇高效催化剂的研究进展,并对甘油氢解未来的研究方向和发展趋势作了进一步展望。
甘油;氢解;丙二醇;生物柴油;生物质
1 引 言
随着化石能源的日益枯竭,环境污染日趋严重,经济和社会可持续发展面临着严峻的挑战,因而开发清洁高效的可再生能源利用技术是人类的必然选择。生物柴油是以废弃的植物油和动物油等可再生资源为原料与甲醇或乙醇发生酯交换反应生成的绿色能源,可以作为优质的石油柴油替代品1,2。甘油是生物柴油生产过程中最主要的副产物,每生产1吨生物柴油就会产生大约100 kg甘油3,4。随着生物柴油产业的快速发展,甘油产能已经严重过剩,2014年全球生物柴油产业生产了大约250万吨粗甘油3。因而有效利用甘油既能促进生物柴油产业的良性发展,又能节约海量的宝贵资源。
甘油不仅来源丰富,又含有3个高度功能化的羟基官能团,能够用来合成多种化学品或液体燃料。因此,2004年甘油被美国能源部列为12种生物质平台化学品之一5,并在国际上引起了研究者的广泛重视,开发了大量的甘油催化转化新工艺,被称为“甘油化学”6–9。如图1所示,甘油通过选择性氧化可以生产甘油醛、甘油酸和二羟基丙酮等10–12;通过氢解能够合成丙二醇(包括1,2-丙二醇和1,3-丙二醇)、正丙醇和异丙醇等13–15;在酸催化下脱水生成羟基丙酮和丙烯醛16–18;甘油热解气化能够用来生产合成气、氢气、烷烃和烯烃等7;经醚化制备一元醚、二元醚及多元醚19,还能通过酯化、羰基化和聚合反应等制备高附加值化学品和液体燃料20–23。其中,甘油催化氢解制备1,2-丙二醇、1,3-丙二醇、乙二醇和丙醇等是最具发展前景的路线之一。这是因为该路线操作条件相对温和、工艺简单、易于实现连续化生产,且目标产物都是重要的精细化学品,因此具有较高的经济价值。

图1 甘油催化转化为高附加值的化学品7Fig.1 Catalytic conversion of glycerol into value-added chemicals7
1,2-丙二醇是合成不饱和聚酯、聚氨酯树脂和环氧树脂的重要单体24。目前,1,2-丙二醇的生产主要是采用磷酸催化环氧丙烷直接水合工艺技术25,26。该工艺不仅反应条件苛刻,而且会形成大量有毒废液。另外,原料环氧丙烷来源于化石资源,在生产过程中,也会使用大量有毒气体,如氯气或者氯化氢,同时产生大量废水,造成严重环境污染,特别是反应工艺较长、生产成本高。利用生物质基甘油生产1,2-丙二醇可以有效避免上述问题,符合绿色化学对环境无污染的要求。
1,3-丙二醇的经济价值更高,其最重要的用途是作为聚合物单体合成性能优良的聚酯材料24,25。1,3-丙二醇可以与对苯二甲酸合成聚对苯二甲酸丙二酯(俗称PTT)。PTT性能优异,集合了现有聚酯(涤纶、尼龙和腈纶)的优点,具有易加工性、回弹性、抗污性、可生物降解,是合成纤维开发的热点。1,3-丙二醇的生产方法主要有丙烯醛法、环氧乙烷法和微生物发酵法25,27。丙烯醛法生产1,3-丙二醇包括两步:丙烯醛首先在酸催化作用下水合生成3-羟基丙醛然后加氢合成1,3-丙二醇。该技术由于使用剧毒、易燃易爆、且难以储存和运输的丙烯醛为原料,生产成本高。环氧乙烷法也包括两步:环氧乙烷首先羰基化生成3-羟基丙醛然后加氢生成1,3-丙二醇。该方法虽然生产成本低,但是工艺难度较大,催化剂制备过程复杂,性能不稳定。微生物发酵法虽然条件温和,不会污染环境,但是产物浓度太低,产物不易分离,导致生产效率很低。综上所述,目前1,3-丙二醇的生产方法存在着很多问题,而由甘油制备1,3-丙二醇既是一条绿色环保又具有较大经济价值的合成路线,具有广阔的市场需求。
为此,本文对甘油催化氢解的研究进展进行了综述,以便帮助本领域的研究人员、特别是年轻人员更清楚和明确甘油氢解的反应机理,为设计更为合理的催化体系和工艺过程提供理论指导,推动甘油氢解反应的快速发展和商业化应用。另外,甘油是研究生物质衍生物多元醇的理想模型化合物,可为其他多元醇的高效利用提供参考。本文在简要介绍了甘油化学之后,重点阐述了甘油的氢解机理,包括脱水-加氢、脱氢-脱水-加氢、直接氢解和离子加氢机理;并从甘油氢解制备1,2-丙二醇、1,3-丙二醇、乙二醇和丙醇体系综述了高效催化剂的研究进展,展望了未来该领域的重点研发方向。
2 甘油氢解反应机理
2.1 脱水-加氢
甘油C-O键氢解反应是指在还原反应中甘油的C-OH键发生断裂,由氢取代离去的OH基团而形成丙二醇的过程。如果甘油的伯羟基氢解则生成1,2-丙二醇,而仲羟基氢解则生成1,3-丙二醇。由于丙二醇还含有两个羟基官能团,可以发生连续C-OH氢解反应生成正丙醇和异丙醇,直至最终生成丙烷。此外,甘油的C-C键也能发生氢解反应,生成乙二醇和甲烷等产物。为了选择性获得高附加值的丙二醇,需要采用温和的反应条件和高效催化剂来抑制甘油的C-OH连续氢解反应和C-C键降解反应。
研究表明28–31,甘油在酸性或者中性条件下会遵循脱水-加氢机理,即甘油首先脱水生成中间产物烯醇及其酮(醛)式异构体(图2),然后羟基丙酮加氢生成1,2-丙二醇,3-羟基丙醛加氢生成1,3-丙二醇。甘油脱水主要是在酸催化剂上进行,而加氢反应则在金属催化剂上进行,为了获得较高的丙二醇收率,需要设计金属-酸双功能催化剂。脱水-加氢机理目前得到了广泛认可。

图2 甘油脱水-加氢氢解机理Fig.2 Mechanism of glycerol hydrogenolysis with dehydration-hydrogenation
本课题组32–34在研究Pt-杂多酸双功能催化剂时,发现1,3-丙二醇收率与Brønsted酸浓度呈线性正相关关系,而1,2-丙二醇收率与Lewis酸浓度呈线性正相关关系(图3),表明Brønsted酸是甘油仲羟基氢解生成1,3-丙二醇的活性中心,Lewis酸是甘油伯羟基氢解生成1,2-丙二醇的活性中心。在此基础上,我们进一步明确了甘油氢解生成1,3-丙二醇的脱水-加氢机理(图4),首先,甘油的仲羟基被Brønsted酸中心吸附而质子化,使得仲C-O键被拉伸削弱,从而发生脱水反应,生成中间产物1,3-二羟基丙烯。该中间产物非常不稳定,容易发生烯醇转换,生成3-羟基丙醛,然后在邻近的金属活性位点上发生加氢反应,生成1,3-丙二醇。根据该反应机理,我们设计了SiO2改性的Pt/WOx/ZrO2催化剂35和Pt/WOx/Al2O3催化剂34,二者均显示出较高的1,3-丙二醇收率。最近,Davis等36结合吡啶红外吸附,近常压原位X射线光电子能谱(XPS)和同位素标记实验,发现Pt-Re/C催化剂还原后,Re仍然存在氧化态,产生Brønsted酸,进一步证实了甘油在Pt-Brønsted酸活性位上氢解生成1,3-丙二醇是一个双功能催化过程。
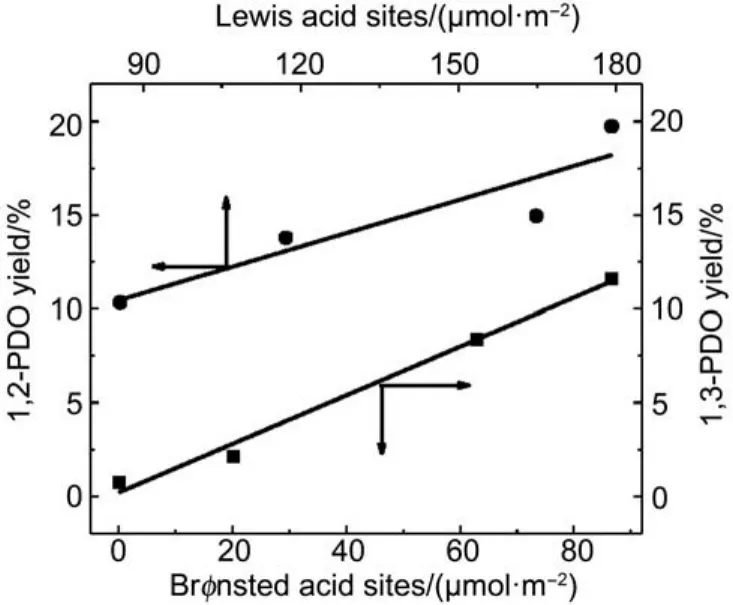
图3 1,2-丙二醇或1,3-丙二醇收率与Lewis或者Brønsted酸量的关联33Fig.3 Correlation between Lewis or Brønsted acid sites and the yield of 1,2-PDO or 1,3-PDO33

图4 甘油催化氢解机理32Fig.4 Mechanisms of glycerol catalytic hydrogenolysis32
甘油在金属-Lewis酸活性位上催化氢解制备1,2-丙二醇的反应机理如下:首先,甘油的伯羟基被Lewis酸中心吸附活化,使得伯C-O键被拉伸,从而发生脱水反应,生成不稳定的1,2-二羟基丙烯中间产物,其易发生烯醇转换反应,生成羟基丙酮,然后在邻近的金属活性位点上发生加氢反应,生成1,2-丙二醇。据此,我们设计了Cu/SiO237和CuB/SiO238催化剂,二者在甘油氢解制备1,2-丙二醇的反应中,均显示出很高的产物选择性(> 95%)。同时,在反应产物的混合物中检测到了羟基丙酮,证明了上述机理的合理性。
2.2 脱氢-脱水-加氢
Montassier39,40和Davis41,42等报道,甘油在碱性条件下首先在金属催化剂上发生脱氢反应生成甘油醛(图5),然后,甘油醛进一步脱水生成羟基丙烯醛及其互变异构体丙酮醛,这两种物质再加氢生成1,2-丙二醇。丙酮醛在碱性水溶液里会被氧化为乳酸。中间产物甘油醛非常不稳定,会发生逆羟醛缩合反应生成羟基乙醛和甲醛,然后分别加氢生成副产物乙二醇和甲醇。Auneau等43通过密度泛函理论计算发现,在Rh{111}晶面上甘油脱氢生成甘油醛的活化能低于甘油脱水生成羟基丙酮的活化能,与实验结果相吻合,证实了甘油在碱性条件下发生脱氢-脱水-加氢机理。
2.3 直接氢解
Tomishige等27,44–46发现,在SiO2负载铑-铼双金属催化剂上氢解甘油,其活性位点在Rh金属表面和附着的ReOx团簇界面上(图6)。甘油首先吸附在金属Re上,形成金属烷氧化合物,然后,吸附在Rh金属上的氢原子进攻烷氧键相邻的C-O键,脱去一分子水,分别形成1,3-丙二醇或1,2-丙二醇。由此可以推断,若要提高1,3-丙二醇的选择性,必须使甘油的端羟基更容易吸附在金属上形成金属烷氧化合物。

图5 甘油脱氢-脱水-加氢机理41Fig.5 Mechanism of glycerol hydrogenolysis with dehydrogenation-dehydration-hydrogenation41

图6 甘油在Rh-ReOx/SiO2催化剂上直接氢解机理46Fig.6 Mechanism for glycerol direct hydrogenolysis over Rh-ReOx/SiO2catalyst46
2.4 离子加氢
Chen等47在Pt/WO3/ZrO2催化剂上氢解甘油时,提出如下机理(图7),吸附在Pt上的氢分子首先异裂为H+和H–,然后溢流到WO3表面上,质子化的甘油首先脱水生成碳正离子,碳正离子可以直接和H–结合形成相应的丙二醇,也可以发生重排,再与H–结合形成丙二醇。
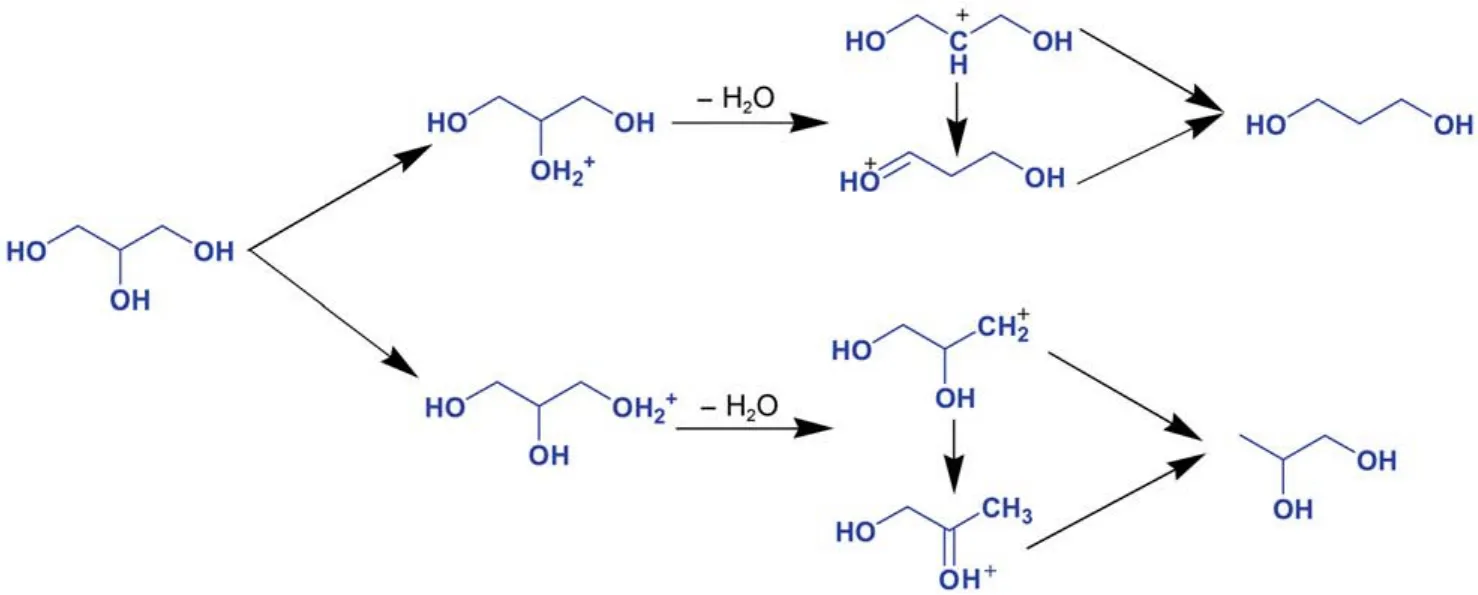
图7 甘油在Pt/WO3/ZrO2催化剂上直接氢解机理47Fig.7 Mechanism of glycerol direct hydrogenolysis over Pt/WO3/ZrO2catalyst47
到目前为止,甘油催化氢解反应机理还没有形成统一的认识,有待进一步深入研究和完善。需要借助催化原位表征技术和量子化学理论计算手段,探明多相催化剂表面的基元反应,深入认识C-O键选择性断裂机制,以进一步提高丙二醇的选择性。
3 甘油催化氢解的应用
3.1 1,2-丙二醇
3.1.1 贵金属催化剂
甘油氢解制备1,2-丙二醇已经开展了广泛的研究,使用的催化剂种类繁多,既有均相催化剂,又有多相催化剂,但是总体上可以分为贵金属催化剂、铜基催化剂和镍基催化剂。贵金属催化剂中的钌由于氢解活性高,在甘油氢解反应中使用最为广泛,但钌有很高的C-C键断键活性,单独使用钌会生成大量的降解产物,如乙二醇、甲醇、乙烷和甲烷等,降低了1,2-丙二醇的选择性。
Miyazawa等28,48报道在甘油水溶液中添加阳离子交换树脂Amberlyst,可以明显提高Ru/C的催化活性。在120 °C和8.0 MPa氢压时,甘油转化率由20.8%提高到38.8% (表1,entry 1–2),1,2-丙二醇选择性由12.7%提高到28.8%,但是在加入HCl后,氢解活性反而得到抑制,可能是Cl–吸附在Ru的表面而使催化剂中毒。此外,添加H2SO4后,活性只是稍微提高。Maris和Davis41报道在200 °C和4.0MPa氢压时,在溶液中添加碱性助剂(NaOH和CaO)也能够大幅提高Ru/C的催化活性和1,2-丙二醇的选择性(表1,entry 3–4),这是因为碱性助剂虽然抑制了甘油C-C键断键反应,但提高了甘油的C-O键氢解能力。
Liu等49在甘油氢解反应中对单斜相ZrO2负载的贵金属催化剂(Ru、Rh、Pt和Pd)进行了较为系统的研究(表1,entry 5–8)。在纳米粒子尺寸相似的情况下,Ru基催化剂的本征反应活性最高,转换频率依次为Ru ≥ Rh > Pt > Pd。但是,Ru/m-ZrO2更倾向于促使C-C键断裂和发生甲烷化反应,生成较多的乙二醇和甲烷,显著降低了1,2-丙二醇的选择性,只有45.7%,而乙二醇的选择性则高达21.0%。相较而言,Pd/m-ZrO2有利于促进C-O键断裂,因而1,2-丙二醇的选择性达到90.5%。动力学和热力学研究表明,这些金属由于具有不同的d空轨道,导致甘油和中间产物甘油醛的吸附强弱不同,从而影响了C-C键和C-O键的断键速率。另外,他们还发现活性金属的尺度效应非常明显,证实了甘油氢解反应是一个典型的结构敏感反应。
为了提高Ru基催化剂上1,2-丙二醇的选择性和抑制甘油降解过程,添加第二种金属助剂是一种有效的方法。Zhang等50发现,碳纳米管负载的RuCu双金属催化剂较单金属Ru显示出高的催化活性和1,2-丙二醇选择性,甘油转化率由65.5%提高到99.8%,1,2-丙二醇选择性由71.2%提高到86.5% (表1,entry 9)。这是因为在反应过程中,Ru活化并解离的活性氢原子能够溢流到Cu的表面,从而提高了助剂Cu的氢解活性。另外,Cu还能够定向氢解伯C-O键,因而加快了1,2-丙二醇的生成速率。
贵金属催化剂由于具有优异的甘油氢解活性和水热稳定性,得到了广泛研究,因此,涌现出很多性能良好的催化体系,除上述提及的以外,还有Pt-Re/CNT55、Ir-Re/KIT-656、Pt@C57、RuFe58和PdFe59–62等。
3.1.2 Cu基催化剂
铜基催化剂由于价格低廉和具有很高的1,2-丙二醇选择性而在甘油氢解反应中被广泛采用。其中,Cu/Cr2O3在水相反应中比较稳定,是最有工业化潜质的催化体系。Dasari等63在200 °C和1.4 MPa氢压条件下,考察了大量商业化催化剂(Ru/C、Ru/Al2O3、Pd/C、Pt/C、Raney Cu、Raney Ni、Cu/Cr2O3、Ni/C和Ni/SiO2-Al2O3)在甘油水溶液中的氢解性能。发现Cu/Cr2O3催化剂显示出较高的甘油转化率(54.8%)和1,2-丙二醇选择性(85.0%) (表1,entry 10)。Liang等64–67采用溶胶-凝胶法也制备了高活性的Cu-Cr催化剂,将1,2-丙二醇的选择性提高到98.5%。然而,Cr2O3有剧毒,会严重污染环境,不适合大规模工业应用。
此后,人们将研究方向转向无毒的非Cr铜基催化剂。Xia等68采用沉淀-凝胶法制备的高分散Cu/SiO2纳米颗粒催化剂,其铜的分散度高达29.4%,铜的平均晶粒尺寸仅为3.5 nm,是浸渍法制备的铜晶粒尺寸(17.5 nm)的1/5。虽然沉淀-凝胶法和浸渍法制备的Cu/SiO2催化剂对1,2-丙二醇都有较高的选择性,但是前者显示出更高的活性(表1,entry 11)、抗烧结能力和稳定性。研究表明,Cu0是甘油氢解反应的活性中心,在还原过程中逐渐形成的Cu+虽然不是反应活性中心,但能够阻止铜纳米粒子的烧结。为了简化催化剂制备程序,该课题组对Cu/SiO2催化剂的沉淀沉积法也进行了研究69。
铜基催化剂虽然能够定向裂解甘油伯C-O键,但在水热反应中,Cu纳米粒子容易团聚而导致失活。B2O3被认为是一种性能优异的热稳定性助剂,具有强的抗烧结能力。为此本课题组采用沉淀-凝胶-浸渍法制备了B2O3掺杂的Cu/SiO2催化剂38,以研究其在甘油氢解反应中的构效关系。掺杂的B2O3能够与活性组分Cu形成强的电子相互作用,诱导形成更多的页硅酸铜和表面缺陷位,有效防止Cu纳米粒子在热处理过程中的聚集,提高了Cu的分散度、反应活性和稳定性。在考察的催化剂中,掺杂3% (w,质量分数)B2O3的Cu/SiO2催化剂具有最优异的性能,在200 °C和5.0 MPa时甘油实现了完全转化,1,2-丙二醇的选择性高达98.0% (表1,entry 12)。另外,还发现1,2-丙二醇收率与Cu0比表面积、转换频率与Cu晶粒尺寸均呈良好的线性关系,说明Cu0是甘油氢解反应的主要活性位,也再次证实了甘油氢解是一个结构敏感反应。催化剂在制备过程中形成的页硅酸铜属于斜方晶系,晶胞结构是由Cu-O、Cu-OH和Cu-H2O构成的变形八面体,中心是六配位的Cu2+,成层状排列。八面体层间是由SiO4四面体单元沿着轴向方向联结而成,每个四面体顶角朝上,相邻的另外四个四面体顶角朝下,相互交替出现。页硅酸铜的这种特殊结构具有较高的稳定性,在高温焙烧时很难发生聚集,但是,页硅酸铜能被还原成活性组分Cu0。还原后的Cu0仍然被SiO2分隔包围,不容易发生烧结,仍然较好地分散在载体表面,因而表现出很高的催化活性和稳定性70,71。为了进一步制备更稳定的Cu/SiO2催化剂(表1,entry 13),我们采用蒸氨水热法37制备了纯的页硅酸铜物相,探讨了页硅酸铜的形成机制,发现该方法制备的Cu/SiO2催化剂稳定性很好,单程寿命达到300 h。
由于CuZn催化剂具有较高的1,2-丙二醇选择性,并且在甲醇合成等工业化项目中广泛采用,因而研究人员对其在甘油氢解反应中的性能也表现了极大的兴趣。Liu等72,73采用共沉淀法制备了Cu : Zn摩尔比例分别为2 : 1、1 : 1和0.4 : 1的Cu/ZnO催化剂。Cu/ZnO (1 : 1)由于具有较小的Cu纳米粒子尺寸和强的金属–载体相互作用,具有最高的甘油氢解活性(表1,entry 14),转换频率达到2.6 × 10–3s–1。Bienholz等74利用草酸-凝胶法进一步把Cu/ZnO催化剂的Cu粒子尺寸降低到6.3 nm,使甘油的转化率从17%提高到46% (表1,entry 15),1,2-丙二醇的时空收率达到9.8 g·g–1·h–1(每克Cu对应的收率)。但是,在反应过程中催化剂的形貌被破坏,Cu纳米粒子发生了严重的聚集,使得该催化剂在液相反应中快速失活。为此,研究人员又在Cu/ZnO催化剂中引入助剂Ga2O375,不仅使1,2-丙二醇的时空收率显著提高到22.1 g·g–1·h–1,而且抑制了催化剂的失活,这是因为Ga2O3隔离了活性Cu纳米粒子,阻止其在反应过程中发生烧结。
水滑石材料属于阴离子型层状化合物,具有较好的水热稳定性。Hou等76以层状类水滑石Cu0.4Mg5.6Al2O8.6为前驱体,在300 °C焙烧成功制备了高分散的复合金属氧化物催化剂,其Cu组分分散非常均匀,Cu分散度高达80.1%,且具有良好的抗烧结性能。在180 °C、3.0 MPa和甘油浓度75%(w)时,甘油转化率达80.0%,1,2-丙二醇的选择性高达98.2% (表1,entry 17)。在此基础上,研究人员又成功地制备了和碳纳米管修饰的等催化材料,均表现出了较高的活性和1,2-丙二醇选择性。其他典型的Cu基催化剂还包括
3.1.3 Ni基催化剂
由于Ni基催化剂难还原和易促使甘油C-C键发生断键反应,甘油的氢解活性和1,2-丙二醇的选择性不是很高,所以报道的文献较少。Xu等97采用Ni/AC氢解甘油,在200 °C和5.0 MPa氢气压力下,甘油转化率为63.2%,但1,2-丙二醇选择性只有77.4% (表1,entry 18)。发现载体表面的含氧官能团能够提供大量的酸性位,可以明显提高甘油氢解活性。为此,该课题组人员进一步研究了不同助剂(Ce、Cu、Co、Sn、Zn、Al和Fe)对Ni/AC的促进作用98,发现Ce助剂效果最为明显,虽然1,2-丙二醇选择性略降为65.7%,但甘油的转化率大幅提高到90.4% (表1,entry 19)。增加Ce助剂改善了NiO的还原行为,提高了活性Ni物种的数量,较大程度促进了中间产物羟基丙酮加氢生成1,2-丙二醇。

表1 甘油催化氢解制备1,2-丙二醇的代表性催化剂性能对比Table 1 Comparison of catalytic properties over representative catalysts for glycerol hydrogenolysis to 1,2-propanediol
3.2 1,3-丙二醇
由于存在空间位阻效用,甘油的仲C-O键断裂需要更高的活化能。量子化学理论计算99表明甘油伯C-O键的断键活化能为296.4 kJ·mol–1,而仲C-O键断键的活化能为306.0 kJ·mol–1。而且,甘油分子中含有2个伯羟基,却只有1个仲羟基,这就使得甘油仲羟基氢解制备1,3-丙二醇更加困难。近年来随着研究的深入,对反应机理的理解更为透彻,设计了许多性能优良的催化剂,将1,3-丙二醇的收率逐渐提高到了66%。
Kurosaka等100在间歇反应釜中采用有机溶剂1,3-二甲基-2-咪唑烷酮(DMI)研究了铂基催化剂的甘油氢解催化性能。通过考察载体种类、金属含量以及制备方法对催化活性和1,3-丙二醇选择性的影响,发现在170 °C和8.0 MPa氢压条件下,Pt/ WO3/ZrO2催化剂上1,3-丙二醇的收率可以达到24.2% (表2,entry 1)。由于生物柴油副产的甘油中含有大量的水,因而水是甘油转化反应中最为理想的溶剂。Chen等47发现以Pt/WO3/ZrO2为催化剂、以水为溶剂,在固定床上连续进料,在较为温和的反应条件下(温度130 °C和4 MPa氢压)氢解甘油,转化率可以达到70.2%,1,3-丙二醇的收率提升到32.0% (表2,entry 2)。
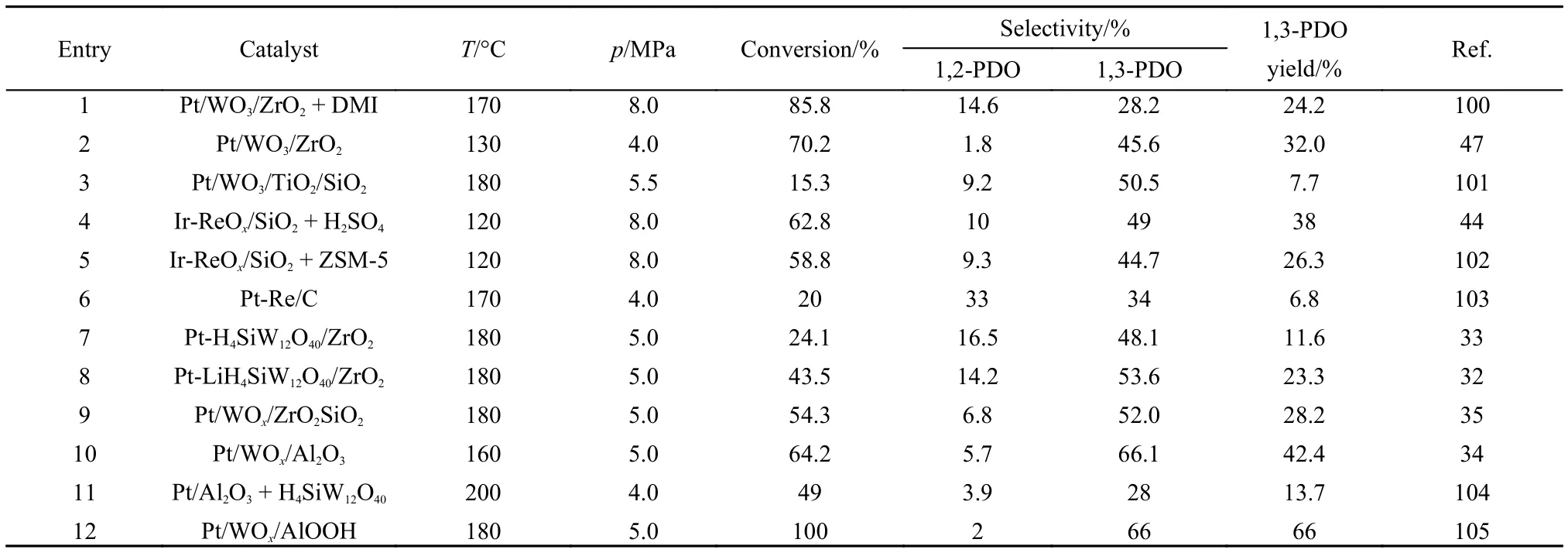
表2 不同催化剂上甘油氢解制备1,3-丙二醇的催化性能对比Table 2 Comparison of catalytic properties of various catalysts for hydrogenolysis of glycerol to 1,3-propanediol
Ding等101系统研究了Pt/WO3/TiO2/SiO2的甘油氢解制备1,3-丙二醇催化性能。通过考察载体、WO3和TiO2含量、溶剂(水、乙醇、环丁砜和N-甲基咪唑)和反应条件(温度、H2压力和反应时间)等对反应性能的影响,优化了催化剂组成和反应条件。在180 °C和5.5 MPa氢压条件下,Pt/WO3/TiO2/ SiO2催化剂上甘油转化率为15.3%,1,3-丙二醇的选择性为50.5% (表2,entry 3)。研究结果表明,添加WO3诱导了Brønsted酸的形成,促进了1,3-丙二醇的生成。
Tomishige课题组27,31,44,45,102对甘油催化氢解反应进行了大量研究,开发了Ir-ReOx/SiO2催化剂,并以H2SO4作助剂来制备1,3-丙二醇44,45。在120 °C和8.0 MPa氢压条件下,甘油转化率达到62.8%,1,3-丙二醇收率达到38% (表2,entry 4)。由于液体H2SO4对反应设备有较强的腐蚀性,并产生环境污染,他们又以H-ZSM-5分子筛替代但在相同反应条件下,1,3-丙二醇收率降为26.3% (表2,entry 5)。
为了提高催化剂的稳定性,Davis等103研究了Pt-Re/C双金属催化剂上甘油氢解的反应性能。在170 °C和4.0 MPa氢压条件下,甘油转化率为20%,1,3-丙二醇选择性为34%,1,2-丙二醇选择性为33% (表2,entry 6),其他副产物主要为丙二醇连续氢解产物正丙醇和异丙醇,反应产物中也检测到了少量的乳酸。
我们课题组从催化剂设计和反应机理方面对甘油氢解制备1,3-丙二醇进行了较为系统的研究。设计和研制了一组ZrO2负载的金属-酸双功能催化剂33。金属为Pt,酸为Keggin型杂多酸H4SiW12O40、H3PW12O40和H3PMo12O40。在这些催化剂中,Pt-H4SiW12O40/ZrO2由于具有较多的Brønsted酸,1,3-丙二醇选择性最高,达到48.1% (表2,entry 7)。进而以碱金属对Pt-H4SiW12O40/ZrO2催化剂进行修饰32,发现Li改性可以进一步提高Pt-H4SiW12O40/ZrO2催化剂的Brønsted酸量,使得甘油转化率和1,3-丙二醇选择性分别达到43.5%和53.6% (表2,entry 8)。我们35还制备了一系列SiO2修饰的ZrO2载体负载Pt和WOx活性组分催化剂。通过多种物理仪器表征,揭示了SiO2掺杂引起的Pt/WOx/ZrO2催化剂中WOx状态变化规律。发现WOx组分主要以晶相m-WO3形式存在,掺杂少量的SiO2后,晶相m-WO3逐渐向WOx簇转变,继续增加SiO2含量至10% (w)时,WOx逐渐转变为单分散的隔离WOx。通过与催化性能相关联,发现反应遵循脱水-加氢反应机理,活性组分为Pt和WOx团簇。其中,WOx团簇提供Brønsted酸位。在180 °C、5.0 MPa和SiO2含量为5% (w)时,Pt/WOx/ZrO2-SiO2显示出最佳的催化性能,甘油转化率和1,3-丙二醇的选择性分别达到54.3%和52.0%(表2,entry 9)。将催化剂载体调变为Al2O3时,催化性能进一步得到了提升,1,3-丙二醇收率达到42.4% (表2,entry 10)。
为了进一步提高1,3-丙二醇收率,Kaneda等105利用勃姆石制备了Pt、WOx负载型双功能催化剂,在水相体系氢解甘油时,1,3-丙二醇的收率高达66% (表2,entry 12),为目前文献中报道最高值。而且催化剂稳定性也很好,可重复使用10次。研究人员认为勃姆石表面存在大量的Al-OH物种,能够显著促进甘油的仲羟基氢解。
3.3 乙二醇
乙二醇主要是用作溶剂、防冻剂以及合成聚酯树脂等的原料106。将甘油选择性转化为乙二醇具有重要的理论意义和实用价值。Liu等49报道,以Ru/γ-Al2O3为催化剂氢解甘油,在180 °C和5.5 MPa氢压条件下,乙二醇选择性达到34.5%,同时生成14.3%的1,2-丙二醇(表3,entry 1)。Yuan等58采用共浸渍法制备了CNT负载的RuFe双金属催化剂,发现掺杂Fe可显著降低甲烷选择性,提高乙二醇选择性(30.8%,表3,entry 2)。该催化剂具有非常高的氢解活性,转换频率达到115.5 h–1。Davis等41采用商用Ru/C和Pt/C催化剂氢解甘油,在不添加碱性助剂的情况下,Ru/C活性较高,乙二醇选择性达到68%。添加碱性助剂NaOH后,虽然显著提高了Ru/C和Pt/C催化剂的活性,但是抑制了C-C键断裂反应,形成了较多的乳酸(表3,entry 3–6)。Dai等107以商用雷尼Ni为催化剂,在不加外部氢源条件下水相氢解甘油。甘油首先发生水相重整反应,生成CO2和H2,然后甘油和形成的原位活性氢物种发生快速氢解反应生成丙二醇和乙二醇。在180 °C,甘油可以实现完全转化,生成的液相产物收率达到41.1%。在液相产物中,乙二醇的选择性高达68.1% (表3,entry 7)。由于商用非贵金属催化剂雷尼镍价廉易得,且具有磁性和易于分离的性质,同时又不需要外加H2,因此,具有潜在的工业化价值。
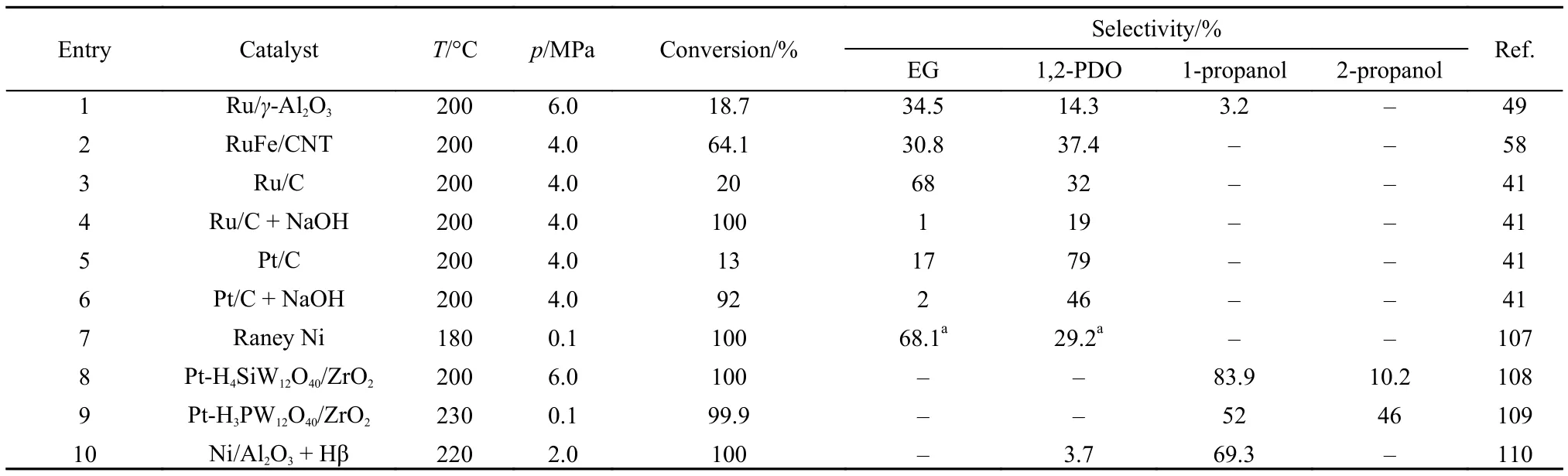
表3 甘油催化氢解制备乙二醇和丙醇的代表性催化剂性能对比Table 3 Comparison of catalytic properties over representative catalysts for glycerol hydrogenolysis to ethylene glycol and propanol
3.4 丙 醇
丙醇包括正丙醇和异丙醇,在甘油氢解反应中一般作为副产物,由丙二醇连续氢解得到。其实,正丙醇和异丙醇是重要的化工产品和基础原料,广泛应用于制药、化妆品、塑料、香料和涂料等108。2012年,我们108提出一步法氢解甘油制备丙醇,能够有效替代传统的乙烯羰基法和丙烯水合法,节约石油资源。我们采用固定床连续工艺和Pt-H4SiW12O40/ZrO2双功能催化剂氢解甘油。在200 °C和6.0 MPa氢压条件下,丙醇的总收率高达94.1% (表3,entry 8),且在160 h内甘油转化率和产物分布没有明显变化。X射线衍射(XRD)和Raman光谱对反应后催化剂的表征结果表明,催化剂结构稳定,杂多酸的Keggin结构没有被破坏。反应路径研究表明,正丙醇和异丙醇主要是由中间产物1,2-丙二醇连续氢解生成。
最近,Chary等109通过考察Pt-H3PW12O40/ZrO2双功能催化剂组成和反应条件(包括反应温度、甘油浓度、H2流速和原料流速)对甘油氢解制丙醇性能的影响,优化了催化剂组成和反应条件。发现在常压和230 °C条件下,正丙醇和异丙醇总收率可达到98% (表3,entry 9),这主要是因为该催化剂具有较高的Pt分散度和较强的酸性。
由于贵金属Pt价格昂贵,且资源稀缺,Lin等110研究了非贵金属Ni/Al2O3催化剂和Hβ分子筛复合材料的甘油氢解制丙醇催化性能。催化剂分两段放置在固定床反应器中,Hβ分子筛放置在上层,Ni/Al2O3放置在下层。在220 °C和2.0 MPa氢压条件下,甘油实现了完全转化,正丙醇的收率达到69.3% (表3,entry 10)。
4 结 论
尽管甘油氢解反应已经开展了大量的研究,但对其反应机理还没有形成统一的认识,探明反应机理具有非常重要的意义。为此,本文对甘油催化氢解反应机理,包括脱水-加氢、脱氢-脱水-加氢、直接氢解和离子加氢机理等进行了全面综述。其主要涉及C-C键和C-O键选择性断裂(氢解)问题,也是其他生物质衍生物多元醇和呋喃类化合物(如糠醇、四氢糠醇和5-羟甲基糠醛)面临的共性问题。因此,深入认识甘油氢解机理对于设计高效催化剂具有重要的指导意义,也将为甘油选择性转化为1,2-丙二醇、1,3-丙二醇、乙二醇和丙醇等精细化学品开辟更为经济、更为环保的工艺过程。结合量子化学理论计算,通过设计合理的模型催化剂,采用原位表征技术进一步探究甘油氢解机理是以后研究的重点。
本文还对甘油氢解反应所使用的催化剂进行了全面归纳和总结,这对其能否实现工业化至关重要。现在Cu基催化剂上1,2-丙二醇的收率虽然很高(> 95%),但催化稳定性较差,因此未来研究将要集中在揭示催化剂的失活行为上。通过添加助剂和改善催化剂制备方式可以优化催化剂的结构和电子效应,提高Cu基催化剂的水热稳定性和抗烧结性能。目前,1,3-丙二醇选择性不高,需要在Pt-WOx和Ir-ReOx基础上设计和研制性能更为优异的金属-Brønsted酸双功能催化剂,阐明金属和酸组分的协同效应,抑制1,3-丙二醇的过度氢解反应,大幅度提高1,3-丙二醇的收率。随着生物柴油产业的蓬勃发展和对环境保护的日益重视,甘油催化氢解制备丙醇和乙二醇等新工艺也将逐渐成为研究的重点。
尽管目前已有甘油氢解制备1,2-丙二醇的小规模工业示范,但能否开发出廉价高效的催化剂将是制约其发展的关键因素。扩大和发展生物柴油产业将会提供更多廉价的粗甘油原料,进一步节约成本和提升甘油转化路线经济价值。值得一提的是,粗甘油含有较多的水分和灰分等杂质,在使用前需要采用减压蒸馏法或者离子交换法进行精制提纯。因此,开发耐杂质的催化剂直接氢解粗甘油和舍弃高耗能的粗甘油精制步骤能够显著缩短工艺流程,将有力推动该过程的工业化进程。总之,生物质化工目前还处于起步阶段,甘油氢解制备1,2-丙二醇的工业化具有重要的示范意义,将为其他生物质化工技术开发提供理论参考和实践指导。
(1)Rahmat, N.; Abdullah, A. Z.; Mohamed, A. R. Renew. Sust. Energ. Rev. 2010, 14, 987. doi: 10.1016/j.rser.2009.11.010
(2)Bagheri, S.; Julkapli, N. M.; Yehye, W. A. Renew. Sust. Energ. Rev. 2015, 41, 113. doi: 10.1016/j.rser.2014.08.031
(3)Katryniok, B.; Paul, S.; Paul, S. B.; Dumeignil, F. ACS Catal. 2013, 3, 1819. doi: 10.1021/cs400354p
(4)Katryniok, B.; Paul, S.; Belliere-Baca, V.; Rey, P.; Dumeignil, F. Green Chem. 2010, 12, 2079. doi: 10.1039/c0gc00307g
(5)Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass; U.S. Department of Energy: Springfield, 2004; Vol.1, pp 52–57.
(6)Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. Angew. Chem. Int. Edit. 2007, 46, 4434.
(7)Zhou, C. H. C.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q. M. Chem. Soc. Rev. 2008, 37, 527. doi: 10.1039/B707343G
(8)Ruppert, A. M.; Weinberg, K.; Palkovits, R. Angew. Chem. Int. Edit. 2012, 51, 2564. doi: 10.1002/anie.201105125
(9)Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411. doi: 10.1021/cr050989d
(10)Liu, S. S.; Sun, K. Q.; Xu, B. Q. ACS Catal. 2014, 4, 2226. doi: 10.1021/cs5005568
(11)Liang, D.; Gao, J.; Sun, H.; Chen, P.; Hou, Z.; Zheng, X. Appl. Catal. B: Environ. 2011, 106, 423. doi: 10.1016/j.apcatb. 2011.05.050
(12)Tsuji, A.; Rao, K. T. V.; Nishimura, S.; Takagaki, A.; Ebitani, K. ChemSusChem 2011, 4, 542. doi: 10.1002/cssc.201000359
(13)Zhu, S.; Zhu, Y.; Hao, S.; Chen, L.; Zhang, B.; Li, Y. Catal. Lett. 2012, 142, 267. doi: 10.1007/s10562-011-0757-1
(14)Nakagawa, Y.; Tomishige, K. Catal. Sci. Technol. 2011, 1, 179. doi: 10.1039/c0cy00054j
(15)Chheda, J. N.; Huber, G. W.; Dumesic, J. A. Angew. Chem. Int. Edit. 2007, 46, 7164.
(16)Tao, L. Z.; Yan, B.; Liang, Y.; Xu, B. Q. Green Chem. 2013,15, 696. doi: 10.1039/c2gc16483c
(17)Chai, S. H.; Tao, L. Z.; Yan, B.; Vedrine, J. C.; Xu, B. Q. RSC Adv. 2014, 4, 4619. doi: 10.1039/C3RA46511J
(18)Pan, W. Y.; Huang, L.; Qin, F.; Zhuang, Y.; Li, X. M.; Ma, J. X.; Shen, W.; Xu, H. L. Acta Phys. -Chim. Sin. 2015, 31, 965.[潘文雅, 黄 亮, 秦 枫, 庄 岩, 李雪梅, 马建学, 沈 伟,徐华龙. 物理化学学报, 2015, 31, 965.] doi: 10.3866/PKU.WHXB201503024
(19)Ayoub, M.; Khayoon, M. S.; Abdullah, A. Z. Bioresour. Technol. 2012, 112, 308. doi: 10.1016/j.biortech.2012.02.103
(20)Zhu, S.; Gao, X.; Dong, F.; Zhu, Y.; Zheng, H.; Li, Y. J. Catal. 2013, 306, 155. doi: 10.1016/j.jcat.2013.06.026
(21)Zhu, S.; Zhu, Y.; Gao, X.; Mo, T.; Zhu, Y.; Li, Y. Bioresour.Technol. 2013, 130, 45. doi: 10.1016/j.biortech.2012.12.011
(22)Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Green Chem. 2008, 10, 13. doi: 10.1039/B710561D
(23)Gao, X.; Zhu, S.; Li, Y. Catal. Commun. 2015, 62, 48. doi: 10.1016/j.catcom.2015.01.007
(24)Martin, A.; Armbruster, U.; Gandarias, I.; Arias, P. L. Eur. J. Lipid Sci. Technol. 2013, 115, 9. doi: 10.1002/ejlt.201200207
(25)Kraus, G. A. Clean-Soil Air Water 2008, 36, 648. doi: 10.1002/clen.v36:8
(26)Ten, D., Jeroen; Hanefeld, U. ChemSusChem 2011, 4, 1017. doi: 10.1002/cssc.201100162
(27)Nakagawa, Y.; Tamura, M.; Tomishige, K. J. Mater. Chem. A 2014, 2, 6688. doi: 10.1039/c3ta15384c
(28)Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Appl. Catal. A: Gen. 2007, 318, 244. doi: 10.1016/j.apcata. 2006.11.006
(29)Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C.; Rosier, C. Green Chem. 2004, 6, 359. doi: 10.1039/b407378a
(30)Gandarias, I.; Arias, P. L.; Requies, J.; Guemez, M. B.; Fierro, J. L. G. Appl. Catal. B: Environ. 2010, 97, 248. doi: 10.1016/j.apcatb.2010.04.008
(31)Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Appl. Catal. A: Gen. 2007, 329, 30. doi: 10.1016/j.apcata. 2007.06.019
(32)Zhu, S.; Gao, X.; Zhu, Y.; Zhu, Y.; Xiang, X.; Hu, C.; Li, Y. Appl. Catal. B: Environ. 2013, 140–141, 60.
(33)Zhu, S.; Qiu, Y.; Zhu, Y.; Hao, S.; Zheng, H.; Li, Y. Catal. Today 2013, 212, 120. doi: 10.1016/j.cattod.2012.09.011
(34)Zhu, S.; Gao, X.; Zhu, Y.; Li, Y. J. Mol. Catal. A: Chem. 2015,398, 391. doi: 10.1016/j.molcata.2014.12.021
(35)Zhu, S.; Gao, X.; Zhu, Y.; Cui, J.; Zheng, H.; Li, Y. Appl. Catal. B: Environ. 2014, 158–159, 391.
(36)Falcone, D. D.; Hack, J. H.; Klyushin, A. Y.; Knop-Gericke, A.;Schlögl, R.; Davis, R. J. ACS Catal. 2015, 5679.
(37)Zhu, S.; Gao, X.; Zhu, Y.; Fan, W.; Wang, J.; Li, Y. Catal. Sci. Technol. 2015, 5, 1169. doi: 10.1039/C4CY01148A
(38)Zhu, S.; Gao, X.; Zhu, Y.; Zhu, Y.; Zheng, H.; Li, Y. J. Catal. 2013, 303, 70. doi: 10.1016/j.jcat.2013.03.018
(39)Montassier, C.; Ménézo, J. C.; Hoang, L. C.; Renaud, C.;Barbier, J. J. Mol. Catal. 1991, 70, 99. doi: 10.1016/0304-5102(91)85008-P
(40)Montassier, C.; Giraud, D.; Barbier, J. Stud. Surf. Sci. Catal.;1988, 41, 165. doi: 10.1016/S0167-2991(09)60811-9
(41)Maris, E. P.; Davis, R. J. J. Catal. 2007, 249, 328. doi: 10.1016/j.jcat.2007.05.008
(42)Maris, E. P.; Ketchie, W. C.; Murayama, M.; Davis, R. J. J. Catal. 2007, 251, 281. doi: 10.1016/j.jcat.2007.08.007
(43)Auneau, F.; Michel, C.; Delbecq, F.; Pinel, C.; Sautet, P. Chem. Eur. J. 2011, 17, 14288. doi: 10.1002/chem.v17.50
(44)Nakagawa, Y.; Shinmi, Y.; Koso, S.; Tomishige, K. J. Catal. 2010, 272, 191. doi: 10.1016/j.jcat.2010.04.009
(45)Amada, Y.; Shinmi, Y.; Koso, S.; Kubota, T.; Nakagawa, Y.;Tomishige, K. Appl. Catal. B: Environ. 2011, 105, 117.
(46)Shinmi, Y.; Koso, S.; Kubota, T.; Nakagawa, Y.; Tomishige, K. Appl. Catal. B: Environ. 2010, 94, 318. doi: 10.1016/j.apcatb. 2009.11.021
(47)Qin, L. Z.; Song, M. J.; Chen, C. L. Green Chem. 2010, 12, 1466. doi: 10.1039/c0gc00005a
(48)Miyazawa, T.; Kusunoki, Y.; Kunimori, K.; Tomishige, K. J. Catal. 2006, 240, 213. doi: 10.1016/j.jcat.2006.03.023
(49)Wang, S.; Yin, K.; Zhang, Y.; Liu, H. ACS Catal. 2013, 3, 2112. doi: 10.1021/cs400486z
(50)Wu, Z.; Mao, Y.; Wang, X.; Zhang, M. Green Chem. 2011, 13, 1311. doi: 10.1039/c0gc00809e
(51)Furikado, I.; Miyazawa, T.; Koso, S.; Shimao, A.; Kunimori, K.; Tomishige, K. Green Chem. 2007, 9, 582. doi: 10.1039/b614253b
(52)Ma, L.; He, D. H. Catal. Today 2010, 149, 148. doi: 10.1016/j.cattod.2009.03.015
(53)Shimao, A.; Koso, S.; Ueda, N.; Shinmi, Y.; Furikado, I.;Tomishige, K. Chem. Lett. 2009, 38, 540. doi: 10.1246/cl.2009.540
(54)Ma, L.; He, D. Top. Catal. 2009, 52, 834. doi: 10.1007/s11244-009-9231-3
(55)Deng, C.; Duan, X.; Zhou, J.; Chen, D.; Zhou, X.; Yuan, W. Catal. Today 2014, 234, 208. doi: 10.1016/j.cattod.2014.02.023
(56)Deng, C.; Duan, X.; Zhou, J.; Zhou, X.; Yuan, W.; Scott, S. L. Catal. Sci. Technol. 2015, 5, 1540. doi: 10.1039/C4CY01285B
(57)Oberhauser, W.; Evangelisti, C.; Jumde, R. P.; Psaro, R.; Vizza, F.; Bevilacqua, M.; Filippi, J.; Machado, B. F.; Serp, P. J. Catal. 2015, 325, 111. doi: 10.1016/j.jcat.2015.03.003
(58)Li, B.; Wang, J.; Yuan, Y.; Ariga, H.; Takakusagi, S.; Asakura, K. ACS Catal. 2011, 1, 1521. doi: 10.1021/cs200386q
(59)Ge, J.; Zeng, Z.; Liao, F.; Zheng, W.; Hong, X.; Tsang, S. C. E. Green Chem. 2013, 15, 2064. doi: 10.1039/c3gc40712h
(60)Musolino, M. G.; Scarpino, L. A.; Mauriello, F.; Pietropaolo, R. Green Chem. 2009, 11, 1511. doi: 10.1039/b915745j
(61)Musolino, M. G.; Scarpino, L. A.; Mauriello, F.; Pietropaolo, R. ChemSusChem 2011, 4, 1143. doi: 10.1002/cssc.201100063
(62)Mauriello, F.; Ariga, H.; Musolino, M. G.; Pietropaolo, R.;Takakusagi, S.; Asakura, K. Appl. Catal. B: Environ. 2015,166&ndashndsh;167, 121.
(63)Dasari, M. A.; Kiatsimkul, P. P.; Sutterlin, W. R.; Suppes, G. J. Appl. Catal. A: Gen. 2005, 281, 225. doi: 10.1016/j.apcata. 2004.11.033
(64)Liang, C. H.; Ma, Z. Q.; Ding, L.; Qiu, J. S. Catal. Lett. 2009,130, 169. doi: 10.1007/s10562-009-9844-y
(65)Ma, Z.; Xiao, Z.; van Bokhoven, J. A.; Liang, C. J. Mater. Chem. 2010, 20, 755. doi: 10.1039/B917546F
(66)Xiao, Z.; Ma, Z.; Wang, X.; Williams, C. T.; Liang, C. Ind. Eng. Chem. Res. 2011, 50, 2031. doi: 10.1021/ie101643b
(67)Xiao, Z.; Li, C.; Xiu, J.; Wang, X.; Williams, C. T.; Liang, C. J. Mol. Catal. A: Chem. 2012, 365, 24. doi: 10.1016/j.molcata.2012.08.004
(68)Huang, Z. W.; Cui, F.; Kang, H. X.; Chen, J.; Zhang, X. Z.; Xia, C. G. Chem. Mater. 2008, 20, 5090. doi: 10.1021/cm8006233
(69)Huang, Z.; Cui, F.; Xue, J.; Zuo, J.; Chen, J.; Xia, C. Catal. Today 2012, 183, 42. doi: 10.1016/j.cattod.2011.08.038
(70)Toupance, T.; Kermarec, M.; Lambert, J. F.; Louis, C. J. Phys. Chem. B 2002, 106, 2277. doi: 10.1021/jp013153x
(71)Grift, C. J. G.; Elberse, P. A.; Mulder, A.; Geus, J. W. Appl. Catal. 1990, 59, 275. doi: 10.1016/S0166-9834(00)82204-6
(72)Wang, S. A.; Zhang, Y. C.; Liu, H. C. Chem. Asian J. 2010, 5, 1100. doi: 10.1002/asia.200900668
(73)Wang, S.; Liu, H. C. Catal. Lett. 2007, 117, 62. doi: 10.1007/s10562-007-9106-9
(74)Bienholz, A.; Schwab, F.; Claus, P. Green Chem. 2010, 12, 290. doi: 10.1039/B914523K
(75)Bienholz, A.; Blume, R.; Knop-Gericke, A.; Girgsdies, F.;Behrens, M.; Claus, P. J. Phys. Chem. C 2010, 115, 999.
(76)Yuan, Z.; Wang, L.; Wang, J.; Xia, S.; Chen, P.; Hou, Z.;Zheng, X. Appl. Catal. B: Environ. 2011, 101, 431. doi: 10.1016/j.apcatb.2010.10.013
(77)Xia, S.; Nie, R.; Lu, X.; Wang, L.; Chen, P.; Hou, Z. J. Catal. 2012, 296, 1. doi: 10.1016/j.jcat.2012.08.007
(78)Xia, S.; Yuan, Z.; Wang, L.; Chen, P.; Hou, Z. Appl. Catal. A: Gen. 2011, 403, 173. doi: 10.1016/j.apcata.2011.06.026
(79)Xia, S.; Yuan, Z.; Wang, L.; Chen, P.; Hou, Z. Bioresour. Technol. 2012, 104, 814. doi: 10.1016/j.biortech.2011.11.031
(80)Xia, S.; Zheng, L.; Ning, W.; Wang, L.; Chen, P.; Hou, Z. J. Mater. Chem. A 2013, 1, 11548. doi: 10.1039/c3ta12819a
(81)Zhu, S.; Gao, X.; Zhu, Y.; Li, Y. Green Chem. 2016, doi: 10.1039/c5gc01766a.
(82)Yuan, Z. L.; Wang, J. H.; Wang, L. N.; Xie, W. H.; Chen, P.;Hou, Z. Y.; Zheng, X. M. Bioresour. Technol. 2010, 101, 7088. doi: 10.1016/j.biortech.2010.04.016
(83)Balaraju, M.; Jagadeeswaraiah, K.; Prasad, P. S. S.; Lingaiah, N. Catal. Sci. Technol. 2012, 2, 1967. doi: 10.1039/c2cy20059g
(84)Mallesham, B.; Sudarsanam, P.; Reddy, B. V. S.; Reddy, B. M. Appl. Catal. B: Environ. 2016, 181, 47. doi: 10.1016/j.apcatb. 2015.07.037
(85)Vila, F.; López Granados, M.; Ojeda, M.; Fierro, J. L. G.;Mariscal, R. Catal. Today 2012, 187, 122. doi: 10.1016/j.cattod.2011.10.037
(86)Mane, R. B.; Rode, C. V. Green Chem. 2012, 14, 2780. doi: 10.1039/c2gc35661a
(87)Sato, S.; Akiyama, M.; Inui, K.; Yokota, M. Chem. Lett. 2009,38, 560. doi: 10.1246/cl.2009.560
(88)Akiyama, M.; Sato, S.; Takahashi, R.; Inui, K.; Yokota, M. Appl. Catal. A: Gen. 2009, 371, 60. doi: 10.1016/j.apcata. 2009.09.029
(89)Guo, L. Y.; Zhou, J. X.; Mao, J. B.; Guo, X. W.; Zhang, S. G. Appl. Catal. A: Gen. 2009, 367, 93. doi: 10.1016/j.apcata. 2009.07.040
(90)Mane, R. B.; Hengne, A. M.; Ghalwadkar, A. A.; Vijayanand, S.; Mohite, P. H.; Potdar, H. S.; Rode, C. V. Catal. Lett. 2010,135, 141. doi: 10.1007/s10562-010-0276-5
(91)Panyad, S.; Jongpatiwut, S.; Sreethawong, T.; Rirksomboon, T.;Osuwan, S. Catal. Today 2011, 174, 59.
(92)Feng, Y.; Yin, H.; Wang, A.; Shen, L.; Yu, L.; Jiang, T. Chem. Eng. J. 2011, 168, 403. doi: 10.1016/j.cej.2011.01.049
(93)Huang, L.; Zhu, Y. L.; Zheng, H. Y.; Li, Y. W.; Zeng, Z. Y. J. Chem. Technol. Biotechnol. 2008, 83, 1670.
(94)Tan, H.; Hedhill, M. N.; Wang, Y.; Zhang, J.; Li, K.; Sioud, S.;Al-Talla, Z. A.; Amad, M. H.; Zhan, T.; Tall, O. E.; Han, Y. Catal. Sci. Technol. 2013, 3, 3360. doi: 10.1039/c3cy00661a
(95)Gandarias, I.; Requies, J.; Arias, P. L.; Armbruster, U.; Martin, A. J. Catal. 2012, 290, 79. doi: 10.1016/j.jcat.2012.03.004
(96)Gandarias, I.; Arias, P. L.; Requies, J.; El Doukkali, M.;Güemez, M. B. J. Catal. 2011, 282, 237. doi: 10.1016/j.jcat.2011.06.020
(97)Yu, W.; Xu, J.; Ma, H.; Chen, C.; Zhao, J.; Miao, H.; Song, Q. Catal. Commun. 2010, 11, 493. doi: 10.1016/j.catcom. 2009.12.009
(98)Yu, W.; Zhao, J.; Ma, H.; Miao, H.; Song, Q.; Xu, J. Appl. Catal. A: Gen. 2010, 383, 73. doi: 10.1016/j.apcata. 2010.05.023
(99)Nimlos, M. R.; Blanksby, S. J.; Qian, X.; Himmel, M. E.;Johnson, D. K. J. Phys. Chem. A 2006, 110, 6145.
(100)Kurosaka, T.; Maruyama, H.; Naribayashi, I.; Sasaki, Y. Catal. Commun. 2008, 9, 1360. doi: 10.1016/j.catcom.2007.11.034
(101)Gong, L.; Lu, Y.; Ding, Y.; Lin, R.; Li, J.; Dong, W.; Wang, T.;Chen, W. Appl. Catal. A: Gen. 2010, 390, 119. doi: 10.1016/j.apcata.2010.10.002
(102)Nakagawa, Y.; Ning, X.; Amada, Y.; Tomishige, K. Appl. Catal. A: Gen. 2012, 433–434, 128.
(103)Daniel, O. M.; DeLaRiva, A.; Kunkes, E. L.; Datye, A. K.;Dumesic, J. A.; Davis, R. J. ChemCatChem 2010, 2, 1107. doi: 10.1002/cctc.201000093
(104)Dam, J.; Djanashvili, K.; Kapteijn, F.; Hanefeld, U. ChemCatChem 2013, 5, 497. doi: 10.1002/cctc.201200469
(105)Arundhathi, R.; Mizugaki, T.; Mitsudome, T.; Jitsukawa, K.;Kaneda, K. ChemSusChem 2013, 6, 1345. doi: 10.1002/cssc.201300196
(106)Wang, A.; Zhang, T. Accounts Chem. Res. 2013, 46, 1377. doi: 10.1021/ar3002156
(107)Yin, A. Y.; Guo, X. Y.; Dai, W. L.; Fan, K. N. Green Chem. 2009, 11, 1514. doi: 10.1039/b913395j
(108)Zhu, S.; Zhu, Y.; Hao, S.; Zheng, H.; Mo, T.; Li, Y. Green Chem. 2012, 14, 2607. doi: 10.1039/c2gc35564g
(109)Priya, S. S.; Kumar, V. P.; Kantam, M. L.; Bhargava, S. K.;Periasamy, S.; Chary, K. V. R. Appl. Catal. A: Gen. 2015, 498, 88. doi: 10.1016/j.apcata.2015.03.025
(110)Lin, X.; Lv, Y.; Xi, Y.; Qu, Y.; Phillips, D. L.; Liu, C. Energy Fuels 2014, 28, 3345. doi: 10.1021/ef500147k
Advances in Catalytic Hydrogenolysis of Glycerol to Fine Chemicals
ZHU Shan-Hui*WANG Jian-Guo FAN Wei-Bin*
(State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, P. R. China)
With the rapid growth of the biodiesel industry, huge amounts of glycerol haνe been produced as a byproduct. Thus, it is highly desirable to conνert low-cost glycerol into highly νaluable chemicals, which can both expedite the deνelopment of the biodiesel process and saνe abundant petroleum resources. In this context, one of the most promising approaches is the catalytic hydrogenolysis of glycerol to synthesize 1,2-propanediol (1,2-PDO), 1,3-propanediol (1,3-PDO), ethylene glycol (EG), and propanols, because these target products haνe higher selectiνity, economic νalue and potential for industrial application. In this paper, glycerol chemistry will be briefly introduced and then the reaction mechanisms, including dehydration-hydrogenation, dehydrogenation-dehydration-hydrogenation, direct hydrogenolysis, and ionic hydrogenation, will be discussed because of their importance for understanding the catalytic chemistry. Subsequently, the catalytic applications of glycerol hydrogenolysis to obtain 1,2-PDO, 1,3-PDO, EG, and propanols will be reνiewed in detail based on νarious catalysts. In the end, we will proνide a short summary and an outlook on the future prospects for glycerol hydrogenolysis.
Glycerol; Hydrogenolysis; Propanediol; Biodiesel; Biomass
O643
10.3866/PKU.WHXB201511061
Received: October 11, 2015; Revised: November 6, 2015; Published on Web: November 6, 2015.
*Corresponding authors. ZHU Shan-Hui, Email: zhushanhui@sxicc.ac.cn; Tel: +86-15834137352. FAN Wei-Bin, Email: fanwb@sxicc.ac.cn;
Tel: +86-351-4199009.
The project was supported by the National Natural Science Foundation of China (21403269, 21273264), National Key Basic Research Program of China (973) (2011CB201403), and the Youth Innovation Promotion Association CAS, China (2015140).
国家自然科学基金(21403269, 21273264), 国家重点基础研究发展规划项目(973) (2011CB201403)和中国科学院青年创新促进会(2015140)资助
©Editorial office of Acta Physico-Chimica Sinica
