木质素催化解聚的研究进展
2016-11-18舒日洋徐莹张琦马隆龙王铁军
舒日洋,徐莹,张琦,马隆龙,王铁军
木质素催化解聚的研究进展
舒日洋1,2,3,徐莹1,2,张琦1,2,马隆龙1,2,王铁军1,2
(1中国科学院可再生能源重点实验室,中国科学院广州能源研究所,广东广州510640;2广东省新能源和可再生能源研究开发与应用重点实验室,广东广州 510640;3中国科学院大学,北京100871)
木质素是一种芳环结构来源丰富且价格低廉的可再生资源。从木质素出发催化解聚制备单酚类高附加值精细化学品和芳香烃烷烃等高品位生物燃料,可以部分替代以化石燃料为原料的生产过程,是生物质资源全组分高效综合利用的重要组成部分。在木质素催化解聚方法中,催化氢解可以直接将木质素转化为低氧含量的液体燃料,在生物燃料利用方面展现出巨大的潜力。本文详细总结了木质素的催化解聚方法,从催化剂类型、溶剂种类、反应机理及催化剂循环使用性等方面介绍了国内外的主要研究进展,着重阐述了木质素催化氢解方法。最后总结了当前木质素催化解聚过程中存在的难题,并对未来的技术发展提出了建议和展望。
生物质;木质素;催化;降解;生物燃料
引 言
木质素是一种主要存在于植物木质部分的复杂高分子化合物,与纤维素和半纤维素一起构成了生物质的三大组分。虽然它的干重只占生物质的10%~35%,但蕴含的能量却占到40%以上[1],因此木质素的转化和利用直接影响生物质的能量利用效率。木质素在自然界含量丰富,且来源广泛,每年从制浆造纸工业蒸煮废液中提取出的工业木质素有近5000万吨,小部分用作建筑材料的添加剂,绝大部分作为廉价燃料烧掉或直接排放,这不仅造成资源浪费而且污染环境[2]。木质素分子是由愈创木基丙烷、紫丁香基丙烷和对羟苯基丙烷3种苯丙烷结构单元随机键合而成的,其中C—O醚键连接方式约占2/3,其余为C—C键,包括β-O-4、α-O-4、4-O-5、β-β、β-5、5-5和β-1键等[3-4]。通过断裂这些连接键,可以得到含有芳香基、甲氧基、酚(醇)羟基、羰基、羧基等多种功能基团的小分子解聚产物。这些小分子化合物C/H含量比与石油相近,在高品质液体燃料和高附加值化学品制备等领域具有极大的应用潜力[3]。
木质素的解聚方法很多,但总体说来利用效率偏低,最关键的问题还是木质素高分子结构的复杂性和坚固性。致密的网状芳环结构和复杂的化学键合方式[3],再加上分子中氢键的作用[5],使得木质素在常见溶剂中溶解性不好甚至不溶,导致解聚难度加大。另外,木质素的多数解聚产物含有醛基、碳碳双键等不饱和官能团,容易发生重聚反应,生成大分子的次级产物,大大降低了解聚效率。在木质素解聚方法中,催化解聚越来越受到人们的关注,催化剂的使用有效降低了连接键的活化能,能够在相对温和的条件下实现连接键的断裂,在木质素解聚利用方面有很大潜力。然而,木质素催化解聚过程非常复杂,催化解聚机理的研究和化学键的演变规律尚不明确,木质素的结构特性、催化剂类型、溶剂种类以及反应条件等对催化解聚效果均有很大影响。其中,木质素的结构特性差异表现在于不同植物来源的木质素有着完全不同的苯丙烷结构单体分布,不同的木质素预处理方法也会导致木质素的相对分子质量和官能团的分布相差很大[1,6];催化剂类型差异在于酸催化剂、碱催化剂和金属催化剂等分别对不同的化学键断裂有特定的催化效果,同时均相催化和异相催化还会展现出完全不同的效果[3];溶剂种类的差异性表现在水、醇类以及其他有机溶剂对不同种类木质素的溶解度不同,这会大大影响原料与催化剂的接触,从而影响催化反应效果;反应条件的差异性体现在温度、反应时间、气氛种类与压力等,能够调控反应速率,加速或抑制副反应的发生。这些因素都会对木质素催化解聚反应效果产生很大影响,得到的产物分布也会随之变化。
弄清木质素的催化解聚机理和化学键的演变规律,认识木质素的内部化学键合方式及其活化与断裂途径,对木质素的高效催化解聚利用具有十分重要的意义。随着科技的发展,越来越多的表征技术开始应用于木质素催化解聚研究。比如,裂解-气相色谱/质谱联用(Py-GC/MS)能通过分析高温裂解木质素产生的小分子可挥发性产物得到木质素分子的元素组分和官能团等特性;傅里叶转换红外线光谱分析(FT-IR)可以通过分子中的化学键或官能团与红外光的振动频率相匹配获得木质素中化学键或官能团的信息;元素分析能通过测量木质素分子的元素组分得到木质素能量密度和推测木质素分子的化学特性;多维核磁共振技术(NMR)可以清晰地看到木质素分子中特定官能团的分布和含量。这些表征技术的应用,让科研工作者对木质素化学键的演变规律及其解聚机理有了更加确切和清晰的认识[7],为木质素的催化解聚的研究和应用奠定了基础。
1 木质素催化解聚方法
木质素的催化解聚方法主要包括生物法和化学法。生物法是利用生物酶作为催化剂实现木质素的解聚,此方法对环境友好、产物选择性高,但解聚速度慢、耗时长、催化剂成本高且易失活[8],因而很难适应木质素的大规模工业应用。相对而言,化学法因其解聚速度快、催化剂价格便宜、对环境要求相对较低而受到青睐。根据木质素催化解聚所采用的催化剂类型、溶剂种类和反应条件的差异,化学法主要分为催化热裂解、催化水解、催化氧化、催化氢解、光催化解聚[9]、电化学解聚[10]等。催化热裂解、催化水解、催化氧化和催化氢解易于实现、经济性好、应用较广,为本文重点介绍的内容。
1.1 催化热裂解
催化热裂解是由早期的普通热裂解发展而来的,具有前处理步骤简便、效率高等优点,适合工业化大规模生产,在国内外受到了广泛的关注[11]。热裂解是在无氧或缺氧条件下,利用热能断开生物质大分子的化学键,发生键的断裂、异构和小分子的聚合等反应,最终转变为低分子量的化学品和燃料[12]。早期的普通热裂解得到的小分子产物收率低,易结焦和积炭[13]。为了提高热解产物中的单酚收率,减少结焦、积炭生成,在木质素的热裂解过程中添加催化剂,可以增加目标产物的选择性和收率。
采用红外线光谱和Py-GC/MS技术分析工业碱木质素的催化热裂解反应发现,溶胶凝胶法制备的TiO2在木质素热解中表现出很高的催化活性,在500℃下可以得到37%(质量)的愈创木酚类产物收率[14],这主要是由于其表面含有大量的羟基和适宜的孔径分布,能够促进催化反应。无定形介孔硅酸铝对木质素催化热裂解也有很好的效果,Al-MCM-41和Al-SBA-15的多孔性有利于产物的扩散,中度酸性则能促进芳烃产物的生成。文献[15]指出,强酸性会加剧积炭,而无定形介孔硅酸铝的中度酸性能减少结焦和积炭的生成。Na2CO3,KCl等无机盐在低温条件下可以提高木质素的失重率,但同时也会促进积炭的生成,而Cu(CH3COO)2和Fe2O3等过渡金属化合物在高温条件下才表现出很高的活性,能够有效提高生物油的收率[16]。甲酸盐同样对木质素催化热裂解有很大的促进作用,在桉木木质素的催化热裂解反应中,温度越高,解聚效果越好。当温度达到300℃,转化率虽然只有65.4%,但生物油产物收率达到61.0%,其中愈创木酚和2,6-二甲氧基苯酚的选择性占到了42.0%[17]。在不同的催化剂下,木质素的热裂解机理和途径也是不同的,对于HZSM-5催化剂,热解油中的酸、醇、醛、酯先裂解为烯烃,然后再发生芳构化反应生成芳基化学品,而CoO/MoO3催化剂则是通过脱除单酚化合物中的甲氧基,直接生成芳香烃化合物[18]。另外,锌催化剂有利于增加自由基的形成[19],氧化铝的存在能够有效催化焦炭中含氧官能团的热分解[20],NaOH有利于木质素中C—C键的断裂[21],钠盐促进木质素的去甲氧基化、去甲基化以及脱氢反应,能够明显改变木质素的热裂解模式[22]。
总体说来,催化热裂解方法简单易于实现,具有很大的工业化潜力。但解聚产物非常复杂,精馏提纯难以实现,产品氧含量高、黏度大,不能作为燃料直接应用于能源终端客户。
1.2 催化水解
酸催化剂作为纤维素和半纤维素水解最常用的催化剂[23],在木质素催化水解上也有相应的应用。从形态上来分,酸催化剂包括液体酸和固体酸。酸性离子液体作为一种典型的液体酸,在早期木质素解聚探索中应用很广,但是离子液体价格昂贵,难以回收利用,废物排放还会污染环境,这些缺点阻碍了它的发展[24]。近年来,甲酸在木质素催化水解上的应用越来越受到人们的关注,它不仅能催化醚键的断裂,促进单酚类化合物的生成,而且还能在解聚过程中分解出氢气为反应过程提供氢源,有助于稳定解聚产物,抑制重聚反应的发生[25]。甲酸对有机溶木质素的水解反应有很好的催化效果,在300℃超临界水条件下,可以得到30%的生物油收率,其中包含了10%~12%的单酚类化合物收率。甲酸的加入还能稳定芳香基团,抑制重聚反应[26]。其他质子酸对木质素水解也有很好的催化作用,碱木质素在以磷酸为催化剂,260℃条件下即可获得46%的液体产物收率和10%左右的单酚类化合物收率[27],文章指出质子酸能有效促进木质素中醚键的断裂,实现解聚。对于固体酸催化剂的研究主要以分子筛为主,弱酸性的SBA-15分子筛对有机溶木质素水解有很好的催化效果,在150℃的低温条件下即可得到以单酚类化合物为主的生物油产物[28]。强酸性的HZSM-5分子筛效果更好,在220℃条件下可以得到15.4%的单酚类化合物收率[29],但是也有文献报道强酸在高温下会促进结焦和积炭的生成[15]。比较其他种类的固体酸催化剂发现,中度酸性的SiO2-Al2O3催化剂对木质素水解也有不错的效果,在250℃条件下可得到30%左右的芳香族单体化合物收率,从催化剂表征、核磁共振技术和红外线光谱分析等结果来看,无定形硅铝催化剂的中度酸性既能满足催化作用的需求,也能有效抑制重聚反应的发生。另外,催化剂具有很好的水热稳定性,表现出优越的循环使用性能,3次重复反应未见明显失活现象[30]。
碱催化剂在木质素催化水解上同样具有不错的效果。常见的碱催化剂主要有LiOH、KOH、NaOH等,具有成本低、来源丰富等优点。木质素水解反应进行时,它们一般溶解在溶剂中实现均相催化。NaOH对木质素水解的催化作用显著,在一定范围内,催化剂碱性越强,原料的转化率越高,芳香族化合物的选择性也越高。强碱能抑制苯环加氢反应的发生,得到苯酚类的目标产物[31]。研究还发现,在NaOH催化木质素水解过程中,添加少量的硼酸和苯酚,对木质素的重聚反应有很强的抑制作用[32]。均相催化虽然效率高,但是很难实现催化剂与产物的分离,废物排放容易污染环境。而固体碱则可以克服这些缺点。MgO固体碱催化剂对有机溶木质素的水解反应有不错的催化效果,在醇与水的混合溶剂下可得到11.2%的单酚类化合物收率,产物与催化剂易于实现分离[33]。
总体说来,均相的酸碱催化剂会与反应溶剂、反应产物互溶,反应结束后难以实现催化剂和反应体系的分离,重复利用难度大、易污染环境。而固体的酸碱催化剂一般不溶于溶剂,反应后与产物分离简单,而且形态和性能相对稳定,更适合工业化应用。
1.3 催化氧化
木质素催化氧化解聚方法能够在温和的条件下得到含多种官能团的芳香族化合物,包括醛类和羧酸类等。根据催化氧化原理的不同,主要分为电化学氧化法、光催化氧化法和化学氧化法。电化学氧化法需要电极来提供氧化反应所需的能量,以Cu-C为电极时,木质素侧链结构被氧化,生成了低分子量的芳基化合物,而且通过调节电量可以改变木质素解聚产物的分子量[34]。研究还发现,加入苯酚化合物有助于提高木质素的氧化降解效率[35]。光催化氧化法则需要在光照条件下进行,紫外光在催化降解高沸醇木质素反应中有很好的效果[36],研究发现光强度和光照时间对木质素的降解起促进作用,降解产物主要包括香草酸、紫丁香基和愈创木基衍生物,其中,香草醛、紫丁香醛、高香草酸等是重要的化石燃料替代品。化学氧化法相对简单,只需加入催化剂来催化氧化,以CuI为催化剂为例,在70℃条件下通过氧化反应和逆Aldol反应的耦合实现了木质素模型化合物的高效转化,最优条件下获得了88%的芳香族单体产物收率[37]。总体说来,所有的催化氧化解聚方法都需要加入氧化剂,O2、H2O2是使用最多的氧化剂,此外还有硝基苯、金属氧化物、二氧化氯和次氯酸盐等。H2O2作为氧化剂对碱木质素解聚有很好的效果,研究发现以水和甲醇为混合溶剂,Fe2(SO4)3为催化剂时木质素的催化氧化解聚效果最好,单酚类产物收率可达到17.9%[38]。另外,固体碱催化木质素解聚的研究中还发现H2O2作为氧化剂时紫丁香基产物居多,而O2作为氧化剂时得到的愈创木基产物较多[39]。对于木质素及其模型化合物的催化氧化研究很多,但深入探究氧化机理的却寥寥无几。值得一提的是,美国威斯康星大学的Stahl[40]研究团队在机理探究方面取得了重大成就。他们先以木质素模型化合物研究了催化氧化反应的路径,发现Cα位置的羟基氧化成酮基对β-O-4键的断裂起决定性作用。然后用真实木质素先氧化后解聚反应,得到61.2%的酚类液体产物,而未氧化的木质素解聚反应只得到7.2%的芳香族化合物收率,文献最后总结了木质素催化氧化反应的规律,提出了详细的反应机理和途径(图1)。
木质素催化氧化解聚过程可显著降低木质素主要化学键断裂的能垒,实现木质素的高效解聚。但是氧化过程中需要添加氧化剂,在促进木质素分子化学键断裂的同时,增加了产物的氧含量,不利于其作为高热值生物燃油的利用。另外,电化学氧化法和光催化氧化法还需要额外的电流和紫外光供给,步骤烦琐,成本较高。
1.4 催化氢解
木质素的催化氢解是指木质素分子在还原反应中醚键断裂,并由氢取代离去的杂原子或基团而后生成小分子产物的过程。木质素催化氢解方法可以直接将木质素转化为低氧含量的液体燃料,具有产物选择性好、热值高、所得产物氧含量低以及显著抑制焦炭等优点,适合工业化发展。木质素催化氢解反应既可以在外加氢气的条件下进行,也可以加入供氢溶剂进行氢转移。从催化剂的类型来看,主要包括贵金属催化氢解、过渡金属催化氢解和双金属催化氢解等。
1.4.1 贵金属催化氢解 贵金属不仅具有优越的吸附氢和解离氢的能力,而且还能催化加氢和氢解反应,因此广泛应用于木质素的催化氢解。其中,负载型贵金属催化剂以其良好的稳定性备受研究者的青睐。一般说来,负载型催化剂是将金属盐类溶液浸渍在载体上,然后经沉淀转化,再加热分解,最后还原制得,可以提高金属组分的分散度和热稳定性,还可以使催化剂有合适的孔结构、形状和机械强度。最常用的贵金属种类主要包括Pd、Pt、Ru、Rh,而载体则主要包括活性炭、固体酸和固体碱。
炭载体具有很大的比表面积,能有效提高贵金属组分的分散度,在木质素氢解上的应用很广。Pd/C催化剂能实现木质素的高效定向氢解,得到了3% 4-乙基苯酚收率,这一收率水平和石油基4-乙基苯酚收率相当[41]。比利时鲁汶大学的Sels团队[42]研究了桦木木质素在Ru/C催化下的氢解效果,在催化剂的作用下,木质素中的醚键容易氢解断裂,生成酚类化合物,产物中含有的不饱和键还会与H2进行加氢反应而饱和,增加产物的稳定性,防止重聚反应的发生。他们还比较了Ru/C和Pd/C下的木质素氢解产物分布的不同,发现Ru/C催化下以愈创木酚类产物为主,而Pd/C催化下则主要生成紫丁香酚类产物[43]。另外,对比研究发现,Pd/C、Rh/C、Ru/C、Pt/C等炭载体催化剂比其他载体催化剂的木质素氢解效果更好[44-45]。
贵金属炭载体催化剂虽然具有很大的比表面积和很高的金属分散度,但是缺少酸碱活性位点对氢解反应的促进作用。而由贵金属炭载体催化剂与酸碱催化剂组成的双功能协同作用催化剂则可以克服这个缺点,对木质素有更好的催化氢解效果。Pd/C和路易斯酸ZnCl2之间的协同催化作用有效促进了木质素模型化合物的催化氢解。研究发现,在温和条件下,催化体系的转化率和目标产物选择性均在80%以上[46]。此催化体系应用到真实木质素的氢解上,同样能实现高效解聚(图2),单酚类化合物收率高达54%[47]。研究还发现,Pd/C和路易斯酸CrCl3同样具有协同催化作用,对碱木质素氢解有很好的催化效果。在最优反应条件下,可得到85.6%的木质素液化率和35.4%的单酚类化合物收率。Pd/C催化剂还具有优越的循环使用性能,重复使用3次,催化效果也只出现稍微下降。红外线光谱分析和扫描电镜分析结果显示催化剂表面有少量积炭生成[48]。另外,该催化剂对脱碱木质素、有机溶木质素、磺酸钠木质素等的氢解均有很好的催化效果[49]。北京大学寇元教授课题组[50]利用Pd/C和固体酸HZSM-5的协同催化作用,实现了木质素模型化合物的高效氢解,得到单酚类产物,再经过进一步加氢脱氧反应,最后制得环烷烃,转化率和产物选择性最高可达100%。他们还发现,Pt/C和磷酸之间的协同催化作用能有效实现桦木木质素的氢解,最高可得到46.4%的单酚类化合物收率和12.0%的二聚体化合物收率[51]。另一方面,金属催化剂与碱催化剂也存在协同作用,有机溶木质素在Ru/C与NaOH的协同催化下,转化率高达92.5%,可以得到12.7%的单酚类化合物和6.1%的脂肪醇类化合物[52]。
直接在固体酸和固体碱载体上负载贵金属也是一种不错的选择,能够有效利用载体的酸碱活性位,对木质素中醚键的氢解断裂起到促进作用。强酸性的Pt/Al2O3负载型催化剂对多种木质素均有催化氢解效果,研究发现强酸性催化剂对β-O-4键的断裂有很好的促进作用,但同时木质素的重聚反应也会加剧[53]。Ru/ZrO2催化剂能有效氢解断裂木质素模型化合物中的β-O-4等醚键,而且,氢气压力对产物分布影响很大,在低氢压条件下,氢解产物可保留苯环的完整性,得到高收率的芳香族碳氢化合物,这为生物航油利用提供了新思路[54]。
1.4.2 过渡金属催化氢解 贵金属催化剂虽然具有不错的催化氢解效果,但是成本太高,而过渡金属则廉价很多,而且同样具备良好的催化氢解性能。最常用的过渡金属主要有Ni,Cu和Fe。
Ni/C催化剂在系列醇溶液中200℃条件下能够实现木质素的定向高效氢解,丙烷基愈创木酚与丙烷基紫丁香酚的选择性高达90%以上。研究发现溶剂对木质素的溶解性与氢解效果有很大关系,溶解性差的水和烷烃溶剂氢解效果较差,而溶解性好的醇类溶剂氢解效果普遍较好,尤其以乙二醇效果最突出[55]。另外醇类溶剂还能作为供氢试剂,为木质素氢解提供氢源[56]。具有弱酸性的Ni/SBA-15催化剂对有机溶木质素的氢解有很好的催化效果。相比于单纯的SBA-15分子筛,负载型催化剂能实现木质素的高效转化,得到更高的生物油收率。其中,负载10% Ni的催化剂效果最好,在30 min反应时间后即可得到30%的生物油产率[57]。另外,在碱性MgO载体上负载Ni,用于催化木质素解聚产物的氢解和加氢反应,也有不错的效果;研究发现反应后产物的分子量显著下降,β-O-4等醚键实现了有效氢解断裂,同时加氢反应稳定了解聚产物,抑制了重聚反应的发生[58]。
Cu催化剂则主要是以Cu掺杂的形式制备多金属氧化物(Cu-PMO)催化剂,这种催化剂有效结合了Cu和碱性位点的催化能力,促进了木质素的氢解效率。美国加利福尼亚大学的Ford教授研究小组[59-62]采用Cu-PMO为催化剂,结合甲醇原位产氢的特性,对木质素模型化合物的催化氢解过程进行了详细的研究,推出了反应机理途径,然后以有机溶木质素为原料实现原位氢解,得到了100%的转化率和50%儿茶酚类产物收率,最后还进行了产物的精制提纯。这为木质素高效利用制备液体燃料和高价值化学品提供了一条新的途径。荷兰埃因霍芬理工大学Hensen教授研究团队[63]则系统研究了固体碱催化剂CuMgAlO在不同反应条件下对木质素的催化氢解反应规律(如表1),研究发现溶剂对反应结果影响很大,超临界乙醇作为溶剂比超临界甲醇效果好很多,这是因为超临界乙醇在固体碱催化剂的作用下,与解聚产物的酚羟基进行了烷基化反应,稳定了解聚产物,抑制了重聚反应的发生[64]。研究还发现CuMgAlO催化剂在380℃下催化木质素氢解反应8 h,即可得到60%(质量)单酚收率。催化剂中Ni的加入能显著增加焦炭的生成,降低单酚收率,而催化剂中合适的Al2O3比例可以有效提高单酚收率[65]。另外,温度的增加能有效促进木质素氢解,但同时也加剧了重聚反应的发生和焦炭的生成,反应时间太长也会使重聚反应增多。值得一提的是,CuMgAlO催化剂不仅对Soda木质素(碱木质素)氢解有很好的效果,能得到60%(质量)的单酚收率,而且对Alcell木质素(溶剂型木质素)和Kraft木质素(磺酸盐木质素)也有很好的反应效果,分别得到62%(质量)和86%(质量)的单酚收率,都远远高于其他文献报道的结果。
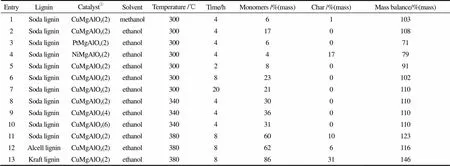
表1 不同反应条件对木质素催化氢解的影响[63-65]
① Cu(Pt/Ni)MgAlOcatalysts all prepared by coprecipitation, loaded with 20%(mass) Cu(Pt/Ni);CuMgAlO(),=(Cu+Mg)/Al, mole.
Fe催化剂在高温下也能表现出加氢和氢解活性。研究发现FeS催化剂对木质素的醚键断裂效果明显,丙三醇作为供氢溶剂,能为木质素氢解提供氢源,在常压下加热到270℃时,即可获得较高的酚类化学品收率。另外FeS作为固体催化剂,不溶于反应溶剂,有利于催化剂的回收利用[66]。
1.4.3 双金属催化氢解 近年来,研究还发现某些特定的双金属催化剂具有协同催化作用,在木质素催化氢解上表现优越。例如NiAu催化剂,在170℃下催化木质素氢解可得到14%以上的单酚收率,比使用单一金属催化剂效果好很多[67]。NiRu催化剂也存在协同催化效果,85%Ni和15%Ru组合的催化剂效果最好,在低温130℃条件下就可以实现木质素的高效转化[68]。
综合来看,木质素催化氢解过程中使用多种功能的催化剂,如金属与酸催化剂协同、金属与碱催化剂协同、金属与金属催化剂协同等,往往比只用一种催化剂的效果更好,这是与木质素复杂的结构特性相关的。一般而言,某一种催化剂一般只对某一类型的反应有明显的催化作用,而木质素的结构复杂,包含各种不同类型的连接方式和官能团,需要多种催化剂协同作用,才能得到高效解聚。然而,其中详细的催化机理途径还有待进一步的研究。
2 结语与展望
综上所述,通过对催化反应体系的合理选择,木质素有望成为高效生产单酚类高附加值精细化学品和芳香烃烷烃类高品位生物燃料的原料,替代不可再生的化石资源。然而要实现木质素催化解聚的工业化利用,还有许多需要完善的地方。
(1)木质素自身结构以及在催化解聚过程中的结构变化需要更深入的认识。虽然近年来科学家们采用多维NMR和高分辨率质谱等新技术较细致地研究了木质素的化学组成和结构特性,但是科学界仍未形成统一和完善的木质素结构理论体系。对于木质素高分子的空间结构,只能近似推导出假设模型。而随着木质素的来源、预处理分离方法和分析检测手段的不同,推出的结构模型也不尽相同,这给木质素催化解聚过程中机理的探究增加了难度。
(2)模型化合物体系的探究与真实木质素的应用相衔接。当前许多的研究多以含β-O-4键的木质素模型化合物为出发点,研究结果与真实体系存在一定的差距。例如,含有β-O-4、α-O-4、4-O-5等醚键的二聚体模型化合物,可以在醇或者水溶剂中通过金属的催化氢解作用有效实现C—O键的断裂,但运用相同或相似的催化体系解聚真实木质素时,却发现木质素的解聚效率非常低。这是因为木质素分子内部虽然以β-O-4等醚键的连接方式居多,但仍存在其他多种相互交错的复杂的化学键合方式,模型化合物虽然具有一定的代表性,但难以全面客观地反映真实原料的转化过程,更无法考察各种连接键、官能团之间的相互影响。因此,做好模型化合物体系和真实木质素体系的衔接工作尤为重要。
(3)针对木质素特殊的结构特性,深入探索高效的催化解聚体系。木质素分子内部致密的网状芳环结构、复杂的化学键合方式以及氢键的作用,使得当前大多数木质素解聚体系存在温度高、反应条件苛刻、时间长、传热传质不畅、催化剂与木质素接触面小等问题,最终导致木质素解聚效率低、产物收率低、易结焦等不足。因此,开发温和、高效、具有普适性的催化体系仍是木质素解聚利用研究的重点。
(4)木质素催化解聚过程中的传热、传质规律需要进一步探究。目前木质素的催化解聚研究,主要集中在工艺条件摸索和催化剂体系的筛选上,而鲜有涉及气、液、固多相复杂流动、混合等传质、热量传递和质能传递-转化的协同促进作用等过程的报道。通过对过程强化的控制,可以促进木质素定向转化,大幅度提高转化效率和降低能耗,实现木质素高效高值化利用,因此,对于过程强化的作用机制还需要深入挖掘。
(5)木质素解聚产物的处理与利用。对木质素催化解聚产物进行分离,实现高价值化学品的利用,是木质素综合利用的重要途径。然而,木质素降解产物成分复杂,性质差异较大,分子量分布参差不齐,很难找到经济合理的分离方法。通过分级萃取和中压制备色谱分离等方法虽然可以获取纯度较高的目标产物,但是操作复杂、成本高、效率低。因此,对于木质素解聚产物的简单高效利用途径,仍有待进一步探索。
References
[1] ZAKZESKI J, BRUIJNINCX P C A, JONGERIUS A L,. The catalytic valorization of lignin for the production of renewable chemicals[J]. Chem. Rev., 2010, 110: 3552-3599.
[2] 孔劼琛, 骆治成, 李愽龙, 等. 木质素解聚和加氢脱氧的进展[J]. 中国科学: 化学, 2015, 45(5):510-525.KONG J C, LUO Z C, LI B L,. Advances in depolymerization and hydrodeoxygenation of lignin[J]. Scientia Sinica: Chimica, 2015, 45:510-525.
[3] 路瑶, 魏贤勇, 宗志敏, 等. 木质素的结构研究与应用[J]. 化学进展, 2013, 25: 839-858. LU Y, WEI Y X, ZONG Z M,. Structural investigation and application of lignins[J]. Progress in Chemistry, 2013, 25: 839-858.
[4] LI C, ZHAO X, WANG A,. Catalytic transformation of lignin for the production of chemicals and fuels[J]. Chem. Rev., 2015, 115(21): 11559-11624.
[5] KUBO S, KADLA J F. Hydrogen bonding in lignin: a Fourier transform infrared model compound study[J]. Biomacromolecules, 2005, 6: 2815-2821.
[6] BRUNOW G. Biorefineries—Industrial Processes and Products[M]. Wiley-VCH Verlag, 2006: 151.
[7] 郭京波, 陶宗娅, 罗学刚. 竹木质素的红外光谱与X射线光电子能谱分析[J]. 化学学报, 2005, 63(16): 1536-1540. GUO J B, TAO Z Y, LUO X G. Analysis of bamboo lignin with FTIR and XPS[J]. Acta Chim. Sinica, 2005, 63(16): 1536-1540.
[8] YAGHOUBI K, PAZOUKI M, SHOJAOSADATI S A. Variable optimization for biopulping of agricultural residues by[J]. Bioresour. Technol., 2008, 99(10): 4321-4328.
[9] LI S H, LIU S Q, COLMENARES J C,. A sustainable approach for lignin valorization by heterogeneous photocatalysis[J]. Green Chem., 2016, 18: 594-607.
[10] CRESTINI C, DAURIA M. Singlet oxygen in the photodegradation of lignin models[J]. Tetrahedron, 1997, 53(23): 7877-7888.
[11] 陈磊, 陈汉平, 陆强, 等. 木质素结构及热解特性[J]. 化工学报, 2014, 65(9): 3626-3633. CHEN L, CHEN H P, LU Q,. Characterization of structure and pyrolysis behavior of lignin[J]. CIESC Journal,2014, 65(9): 3626-3633.
[12] 程辉, 余剑, 姚梅琴, 等. 木质素慢速热裂解机理[J]. 化工学报, 2013, 64(5): 1757-1765. CHENG H, YU J, YAO M Q,. Mechanism analysis of lignin slow pyrolysis[J]. CIESC Journal,2013, 64(5): 1757-1765.
[13] 武书彬, 向冰莲, 刘江燕, 等. 工业碱木素热裂解特性研究[J]. 北京林业大学学报, 2008, 30(5): 143-147. WU S B, XIANG B L, LIU J Y,. Pyrolysis characteristics of technical alkali lignin[J]. Journal of Beijing Forestry University, 2008, 30(5): 143-147.
[14] NAIR V, VINU R. Production of guaiacolscatalytic fast pyrolysis of alkali lignin using titania, zirconia and ceria [J]. J. Anal. Appl. Pyrol., 2016, 119: 31-39.
[15] CUSTODIS V B, KARAKOULIA S A, TRIANTAFYLLIDIS K S,. Catalytic fast pyrolysis of lignin over high-surface-area mesoporous aluminosilicates: effect of porosity and acidity[J]. ChemSusChem, 2016, 9: 1134-1145.
[16] AMEN-CHEN C, PAKDEL H, ROY C. Production of monomeric phenols by thermochemical conversion of biomass[J]. Bioresour. Technol., 2001, 79: 277-299.
[17] SCHUCHARDT U, RODRIGUES J A R, COTRIM A,. Liquefaction of hydrolytic eucalyptus lignin with formate in water, using batch and continuous-flow reactors[J]. Bioresour. Technol., 1993, 44(2): 123-129.
[18] MULLEN C A, BOATENG A A. Catalytic pyrolysis-GC/MS of lignin from several sources[J]. Fuel Process. Technol., 2010, 91: 1446-1458.
[19] SHAFIZADEH F, CHIN P P S. Thermal deterioration of wood[J]. Wood Technol.: Chem. Aspects, Symposium, 1976, 43: 57-81.
[20] SERIO M E, BASSILAKIS R, SOLOMON P R. Production of carbon materials from biomass[J]. Division Fuel Chem., 1991, 36: 1110-1118.
[21] DOMBERGS G, KIRSBAUMS I, DOBELE G,. Effect of alkaline additives on the formation of phenols during lignin pyrolysis[J]. Koksnes Kimija, 1976, 5: 73-80.
[22] KLEEN M, GELLERSTEDT G. Influence of inorganic species on the formation of polysaccharide and lignin degradation products in the analytical pyrolysis of pulps[J]. J. Anal. Appl. Pyrol., 1995, 35: 15-41.
[23] 金强, 张红漫, 徐锐, 等. 半纤维素稀酸循环喷淋冲滤水解动力学[J]. 化工学报, 2011, 62(1): 103-110. JIN Q, ZHANG H M, XU R,. Kinetics of hemicellulose hydrolysis by dilute acid with cycle spray flow-through[J]. CIESC Journal,2011, 62(1): 103-110.
[24] LONG J X, LOU W Y, WANG L F,. [C4H8SO3Hmim]HSO4as an efficient catalyst for direct liquefaction of bagasse lignin: decomposition properties of the inner structural units[J]. Chem. Eng. Sci., 2015, 122: 24-33.
[25] LIGUORI L, BARTH T. Palladium-Nafion SAC-13 catalysed depolymerisation of lignin to phenols in formic acid and water[J]. J. Anal. Appl. Pyrol., 2011, 92(2): 477-484.
[26] GOSSELINK R J, TEUNISSEN W, VAN DAM J E,. Lignin depolymerisation in supercritical carbon dioxide/acetone/water fluid for the production of aromatic chemicals[J]. Bioresour. Technol., 2012, 106: 173-177.
[27] ZHANG X H, ZHANG Q, LONG J X,. Phenolics production through catalytic depolymerization of alkali lignin with metal chlorides[J]. Bioresources, 2014, 9(2): 3347-3360.
[28] TOLEDANO A, SERRANO L, BALU A M,. Fractionation of organosolv lignin from olive tree clippings and its valorization to simple phenolic compounds[J]. ChemSusChem, 2013, 6(3): 529-536.
[29] SINGH S K, EKHE J D. Towards effective lignin conversion: HZSM-5 catalyzed one-pot solvolytic depolymerization/hydrodeoxygenation of lignin into value added compounds[J]. RSC Advances, 2014, 4(53): 27971-27978.
[30] DEEPA A K, DHEPE P L. Lignin depolymerization into aromatic monomers over solid acid catalysts[J]. ACS Catal., 2014, 5: 365-379.
[31] KONNERTH H, ZHANG J G, MA D,. Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water[J]. Chem. Eng. Sci., 2015, 123: 155-163.
[32] TOLEDANO A, SERRANO L, LABIDI J. Improving base catalyzed lignin depolymerization by avoiding lignin repolymerization[J]. Fuel, 2014, 116: 617-624.
[33] LONG J X, ZHANG Q, WANG T J,. An efficient and economical process for lignin depolymerization in biomass-derived solvent tetrahydrofuran[J]. Bioresour. Technol., 2014, 154: 10-17.
[34] KOVALENKO E I, SMIRNOV V A, SHALIMOV V N. Effect of anode material and electrolysis time on the directivity of the electrochemical oxidation of hydrolytic lignin[J]. Zhurnal Prikladnoi Khimii, 1977, 50(8): 1741-1744.
[35] DIAZ-GONZALEZ M, VIDAL T, TZANOV T. Phenolic compounds as enhancers in enzymatic and electrochemical oxidation of veratryl alcohol and lignin[J]. Appl. Microbiol. Biotechnol., 2011, 89: 1693-1700.
[36] 陈婷, 陈云平, 张英, 等. TiO2光催化氧化降解高沸醇木质素的研究[J]. 纤维素科学与技术, 2010, 18(4): 13-18. CHEN T, CHEN Y P, ZHANG Y.. Study on degradation of high boiling solvent lignin by TiO2photochemical catalysis[J]. Cellulose Sci. Technol., 2010, 18(4): 13-18.
[37] ZHANG J, LIU Y, CHIBA S,. Chemical conversion of beta-O-4 lignin linkage models through Cu-catalyzed aerobic amide bond formation[J]. Chem. Commun., 2013, 49: 11439-11441.
[38] OUYANG X P, RUAN T, QIU X Q. Effect of solvent on hydrothermal oxidation depolymerization of lignin for the production of monophenolic compounds[J]. Fuel Process. Technol., 2016, 144: 181-185.
[39] YANG Q, SHI J, LIN L,. Characterization of changes of lignin structure in the processes of cooking with solid alkali and different active oxygen[J]. Bioresour. Technol., 2012, 123: 49-54.
[40] RAHIMI A, ULBRICH A, COON J J,. Formic-acid-induced depolymerization of oxidized lignin to aromatics[J]. Nature, 2014, 515(7526): 249-252.
[41] YE Y Y, ZHANG Y, FAN J,. Selective production of 4-ethylphenolics from ligninmild hydrogenolysis[J]. Bioresour. Technol., 2012, 118: 648-651.
[42] VAN DEN BOSCH S, SCHUTYSER W, VANHOLME R,. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps[J]. Energy Environ. Sci., 2015, 8(6): 1748-1763.
[43] VAN DEN BOSCH S, SCHUTYSER W, KOELEWIJN S F,. Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood[J]. Chem. Commun., 2015, 51: 13158-13161.
[44] PEPPER J M. Lignin and related compounds(I): A comparative study of catalysts for lignin hydrogenolysis[J]. Can. J. Chem., 1969, 47(5): 723-727.
[45] XU W, MILLER S J, AGRAWAL P K,. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid[J]. ChemSusChem, 2012, 5(4): 667-675.
[46] PARSELL T H, OWEN B C, KLEIN I,. Cleavage and hydrodeoxygenation (HDO) of C—O bonds relevant to lignin conversion using Pd/Zn synergistic catalysis[J]. Chem. Sci., 2013, 4(2): 806-813.
[47] PARSELL T, YOHE S, DEGENSTEIN J,. A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass[J]. Green Chem., 2015, 17(3): 1492-1499.
[48] SHU R Y, LONG J X, YUAN Z Q,. Efficient and product-controlled depolymerization of lignin oriented by metal chloride cooperated with Pd/C[J]. Bioresour. Technol., 2015, 179: 84-90.
[49] SHU R Y, LONG J X, XU Y,. Investigation on the structural effect of lignin during the hydrogenolysis process[J]. Bioresour. Technol., 2016, 200: 14-22.
[50] ZHAO C, LERCHER J A. Selective hydrodeoxygenation of lignin-derived phenolic monomers and dimers to cycloalkanes on Pd/C and HZSM-5 catalysts[J]. ChemCatChem, 2012, 4(1): 64-68.
[51] YAN N, ZHAO C, DYSON P J,. Selective degradation of wood lignin over noble-metal catalysts in a two-step process[J]. ChemSusChem, 2008, 1(7): 626-629.
[52] LONG J X, XU Y, WANG T J,. Efficient base-catalyzed decomposition and in situ hydrogenolysis process for lignin depolymerization and char elimination[J]. Appl. Energy, 2015, 141: 70-79.
[53] BOUXIN F P, MCVEIGH A, TRAN F,. Catalytic depolymerisation of isolated lignins to fine chemicals using a Pt/alumina catalyst(part 1): Impact of the lignin structure[J]. Green Chem., 2015, 17(2): 1235-1242.
[54] LUO Z C, WANG Y M, HE M Y,. Precise oxygen scission of lignin derived aryl ethers to quantitatively produce aromatic hydrocarbons in water[J]. Green Chem., 2016, 18: 433-441.
[55] SONG Q, WANG F, XU J. Hydrogenolysis of lignosulfonate into phenols over heterogeneous nickel catalysts[J]. Chem. Commun., 2012, 48(56): 7019-7021.
[56] SONG Q, WANG F, ZHANG J J,. Lignin depolymerization (LDP) in alcohol over nickel-based catalystsa fragmentation–hydrogenolysis process[J]. Energy Environ. Sci., 2013, 6(3): 994-1007.
[57] TOLEDANO A, SERRANO L, PINEDA A,. Microwave-assisted depolymerisation of organosolv ligninmild hydrogen-free hydrogenolysis: catalyst screening[J]. Appl. Catal. B: Environ., 2014, 145: 43-55.
[58] LONG J X, SHU R Y, YUAN Z Q,. Efficient valorization of lignin depolymerization products in the present of NiMg1−xO[J]. Appl. Energy, 2015, 157: 540-545.
[59] MACALA G S, MATSON T D, JOHNSON C L,. Hydrogen transfer from supercritical methanol over a solid base catalyst: a model for lignin depolymerization[J]. ChemSusChem, 2009, 2(3): 215-217.
[60] BARTA K, MATSON T D, FETTIG M L,. Catalytic disassembly of an organosolv ligninhydrogen transfer from supercritical methanol[J]. Green Chem., 2010, 12(9): 1640-1647.
[61] BARTA K, FORD P C. Catalytic conversion of nonfood woody biomass solids to organic liquids[J]. Accounts Chem. Res., 2014, 47(5): 1503-1512.
[62] BARTA K, WARNER G, BEACH E S,. Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides[J]. Green Chem., 2014, 16: 191-196.
[63] HUANG X M, KORANYI T I, BOOT M D,.Catalytic depolymerization of lignin in supercritical ethanol[J]. ChemSusChem, 2014, 7(8): 2276-2288.
[64] HUANG X M, KORANYI T I, BOOT M D,. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics[J]. Green Chem., 2015, 17: 4941-4950.
[65] HUANG X M, ATAY C, KORANYI T I,. Role of Cu-Mg-Al mixed oxide catalysts in lignin depolymerization in supercritical ethanol[J]. ACS Catal., 2015, 5(12): 7359-7370.
[66] 隋鑫金, 武书彬. 工业碱木素热化学转化制备酚类化学品[J]. 化工学报, 2011, 62(6): 1763-1769. SUI X J, WU S B. Preparation of phenols using thermal chemical conversion of industrial kraft lignin [J]. CIESC Journal, 2011, 62(6): 1763-1769.
[67] ZHANG J G, ASAKURA H, VAN RIJN J,. Highly efficient, NiAu-catalyzed hydrogenolysis of lignin into phenolic chemicals[J]. Green Chem., 2014, 16(5): 2432-2437.
[68] ZHANG J G, TEO J, CHEN X,. A series of NiM (M = Ru, Rh, and Pd) bimetallic catalysts for effective lignin hydrogenolysis in water[J]. ACS Catalysis, 2014, 4(5): 1574-1583.
Progress in catalytic depolymerization of lignin
SHU Riyang1,2,3, XU Ying1,2, ZHANG Qi1,2, MA Longlong1,2, WANG Tiejun1,2
(1Key Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, Guangdong, China;2Guangdong Key Laboratory of New and Renewable Energy Research and Development, Guangzhou 510640, Guangdong, China;3University of Chinese Academy of Sciences, Beijing 100871, China)
Lignin is a cheap and renewable resource with rich aromatic units. It can be efficiently transformed into highly value-added fine chemicals such as phenolic monomers and other high-grade biofuels such as arenes and alkanes through catalytic depolymerization methods, which has long been regarded as an important constituent part of biomass resources comprehensive utilization approaches. This process is able to replace the chemicals production from the fossil fuel partly. Among the lignin depolymerization methods, the catalytic hydrogenolysis process can directly convert lignin into liquid fuel with low oxygen contents, and these biofuels show a great potential in the replacement of traditional energy sources. This paper focuses on the catalytic lignin depolymerization methods. The recent progress at home and abroad is reviewed based on the catalysts, solvents, catalysis mechanism and catalyst recyclability. Wherein, the catalytic hydrogenolysis method is emphatically introduced in detail. Furthermore, the current technique challenges during the lignin catalytic depolymerization process are summarized. Many future technologic explorations and suggestions for the efficient application of lignin are proposed.
biomass; lignin; catalysis; degradation; biofuel
2016-06-29.
Prof. ZHANG Qi, zhangqi@ms.giec.ac.cn
10.11949/j.issn.0438-1157.20160885
TK 6
A
0438—1157(2016)11—4523—10
舒日洋(1990—),男,博士研究生。
国家科技支撑项目(2014BAD02B01);国家自然科学基金项目(51476178)。
2016-06-29收到初稿,2016-08-23收到修改稿。
联系人:张琦。
supported by the National Key Technology R&D Program (2014BAD02B01) and the National Natural Science Foundation of China (51476178).
