Pharmacogenomics of Drug Metabolizing Enzymes and Transporters:Relevance to Precision Medicine
2016-11-09ShabbirAhmedZhanZhouJieZhouShuQingChen
Shabbir AhmedZhan ZhouJie ZhouShu-Qing Chen*d
1Department of Precision Medicine and Biopharmaceutics,College of Pharmaceutical Sciences,Zhejiang University,Hangzhou 310058,China
2International Center for Precision Medicine,Zhejiang California International NanoSystems Institute,Hangzhou 310058,China
REVIEW
Pharmacogenomics of Drug Metabolizing Enzymes and Transporters:Relevance to Precision Medicine
Shabbir Ahmed1,a,Zhan Zhou1,b,Jie Zhou1,c,Shu-Qing Chen1,2,*,d
1Department of Precision Medicine and Biopharmaceutics,College of Pharmaceutical Sciences,Zhejiang University,Hangzhou 310058,China
2International Center for Precision Medicine,Zhejiang California International NanoSystems Institute,Hangzhou 310058,China
Pharmacogenomics;
Precision medicine;
Genetic polymorphism;
Phase-I drug-metabolizing enzymes;
Drug transporters
The interindividual genetic variations in drug metabolizing enzymes and transporters influence the efficacy and toxicity of numerous drugs.As a fundamental element in precision medicine,pharmacogenomics,the study of responses of individuals to medication based on their genomic information,enables the evaluation of some specific genetic variants responsible for an individual's particular drug response.In this article,we review the contributions of genetic polymorphisms to major individual variations in drug pharmacotherapy,focusing specifically on the pharmacogenomics of phase-I drug metabolizing enzymes and transporters.Substantial frequency differences in key variants of drug metabolizing enzymes and transporters,as well as their possible functional consequences,have also been discussed across geographic regions.The current effort illustrates the common presence of variability in drug responses among individuals and across all geographic regions.This information will aid health-care professionals in prescribing the most appropriate treatment aimed at achieving the best possible beneficial outcomes while avoiding unwanted effects for a particular patient.
Introduction
aORCID:0000-0003-0672-1290.
bORCID:0000-0002-2730-5483.
cORCID:0000-0003-2176-6366.
dORCID:0000-0002-0792-3735.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.03.008
1672-0229Ⓒ2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
Pharmacogenomics is the understanding of how individuals differ in their response to drug therapy and the mechanisms underlying variable drug response by utilizing genomics,proteomics,transcriptomics,and metabolomics based knowledge. Every individual has a different genetic makeup,which influences the risk of developing diseases as well as responses to drugs and environmental factors[1].Genomic differences between individuals are present approximately every 300-1000 nucleotides with over 14 million single nucleotide polymorphisms(SNPs)distributed throughout the entire humangenome[2].Therefore,identification of DNA variants that most significantly contribute to the population variations in each trait is one of the fundamental objectives of genetics[3]. The understanding of variations in interindividual drug response behaviors has been greatly improved owing to the rapid developments in pharmacogenomics over the last few years.Each individual in a large patient population responds differently,which possibly explains why a treatment that has been proven efficacious in some patients often fails to elicit adequate responses in others.Moreover,such treatment failure in the affected patients may cause some serious side effects or even lead to death,which is inductive of individual variability in drug safety and efficacy.The causative factors for variations in drug response are complex and multifold with direct or indirect consequences.Among them,stably-inherited genetic factors are the major variables[4],whereas others include environmental factors like chemicals and radiation exposure,lifestyle factors like drinking,smoking and exercise,and physiological factors like age,sex,liver and kidney function,pregnancy,and starvation[5].It is evident from previous studies that population variability in drug response is often larger than intrapatient variability(within the same individual at different time points)[6].
Drug response of individual patients is primarily determined by the pharmacokinetic and pharmacodynamic properties of prescribed drugs,which is directly or indirectly affected by polymorphisms in drug metabolizing enzymes and transporters.Different populations have varied allele frequencies in genes of both drug metabolizing enzymes and transporters. For precision medicine,the molecular and clinical information is integrated in order to understand the biological basis of disease and develop medications with better outcomes for patients[7].Therefore,precision medicine will help to improve the selection of disease targets and lead to the identification of patient populations that exhibit better clinical result at normal doses[8].
Variations in drug response
It is well known that individuals vary significantly in their clinical responses to administered drugs and the outcomes,which can be inherited or acquired,are always patient-specific[9]. Such interindividual variation is often a challenge to optimizing a dosage regimen because most drugs are effective in only 25%-60%of patients[10].Many patients are unable to fully respond and benefit from the first recommended drug treatment.For example,an average of 38%,40%,43%,50%,and 75%of patients who have depression,asthma,diabetes,arthritis,and cancer,respectively,show no response to initial treatments[11].
Different patients can respond differently to the same drug and dose.Sometimes,the effective drug dose for a particular patient may prove lethal to or result in therapeutic failure in others(too low drug concentrations at normal doses),leading to serious adverse effects or no effects at all.Continuous drug monitoring is recommended when prescribing drugs with known serious side effects and narrow therapeutic indexes to avoid unexpected and undesirable outcomes[12].The situation can worsen if the patient takes other drugs and has other existing disease conditions due to possible drug-drug and drug-disease interactions[13].For example,the daily warfarin dose varies by up to 20-to 30-fold between patients in many disease conditions where it is recommended for the treatment of embolism and thrombosis[13].Similar observation has also been reported for dose-dependent individual variations in drug response to simvastatin,an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase(HMGCR)[14].
The recommended daily maximum dose of simvastatin for the management of blood cholesterol levels is 40 mg.In a cohort study of 156 patients,95%of them showed reduced levels of low-density lipoprotein(LDL)cholesterol,whereas the remaining 5%exhibited no reduction was observed for the remaining 5%of the patients,even at doses as high as 160 mg/day of simvastatin[15].It is suggested that the genetic polymorphisms in genes encoding ATP-binding cassette subfamily G member 2(ABCG2)and HMGCR contribute to the interindividual difference in a dose-dependent manner[14,16].
Contributing factors in interindividual drug responses
Individual-specific response to medication can be attributed to many multifold and complex factors including the unique genetic makeup(mutations such as SNPs,gene deletions,and duplications).These genetic factors,as well as physiological conditions(age,gender,body size,and ethnicity);environmental influences(exposure to toxins,diet,and smoking);and pathological factors(liver and renal function,diabetes,and obesity)can work alone or in combination to influence drug responses[17].According to the hypothesis of Tang et al.[18],various genetic factors contribute approximately 20%-95%to determining the interindividual variability in drug responses.Furthermore,individual variations in responses related to genetic factors are often permanent,while those influenced by other factors are mostly transient[6].In support of inheritance being a major determinant of drug response,Vesell et al.[19]found relatively higher population variability of a drug response among all the individuals in a population than the intrapatient variability at different times.
Determinants of interindividual drug responses
Disease conditions of individuals used to be diagnosed based on signs and symptoms,which may be indicative of several different diseases or somewhat related to the family history.In the past,clinicians could only attempt to cure or treat disease upon its onset[20].Currently,more specific and precise diagnostic approaches have been developed to examine genes and the genetic variants known to be associated with altered interindividual drug response or specific diseased conditions. Success of the Human Genome Project(HGP)has contributed considerably in this context.Pharmacogenomics enables scientists to assess specific genetic variants that may be responsible for an individual's particular drug response by identifying the particular genetic loci involved[21].Whole-genome SNP profiling,haplotyping,multigene analysis,and gene expression studies using biochips or microarrays[22,23]are recently used to study individual responses to drugs at various levels and could facilitate drug discovery and development[24].
Genetic polymorphisms may influence a drug's effect by altering its pharmacokinetics,pharmacodynamics,or both(Figure 1),which are two major determinants conferring the interindividual differences in drug responses.Pharmacokinetics deals with how much of a drug is required to reach its target site in the body,while pharmacodynamics deals with how well the targets such as receptors,ion channels,and enzymes respond to various drugs[25,26].Genetic polymorphisms in drug transporters and phase-1 drug-metabolizing enzymes can alter the pharmacokinetic and pharmacokinetic properties of the administered drugs,their metabolites or both at the target site,resulting in variability in drug responses.Theoretically,variations at even a single base(SNPs)or sets of closely-related SNPs(haplotypes)in genes involved in the pharmacokinetic and pharmacodynamic pathways at any stage could affect the overall drug response of an individual[27,28].
Mutationsinthegenecodingregionscouldcause alterations in gene expression or protein structure,leading to variations in protein quantity and quality.In the case of enzymes,such mutations affect both the protein function and the rate and kinetic constants.Changes in drug-receptor or drug-enzyme interactions due to structural alterations of enzymes or receptors could also result in variations in drug responses[6].Polymorphisms in genes responsible for drug transport can affect pharmacokinetic properties of an administered drug and ultimately its plasma concentration as well as concentrations in the target tissues.In addition,altered drug response could also be attributed to reduced repairing capability for mutations triggered by alkylating agents due to malfunctioning of DNA repair enzymes[29].Such protective effect could be affected by genetic polymorphisms causing altered protein structure or reduced expression in enzymes responsible for glutathione biosynthesis[2].
Twin studies have provided evidence supporting the contribution of genetic factors to individuals'varied drug responses. For instance,in the late 1950s,it was found that dizygotic twins exhibited more metabolic variability than did monozygotic twins for isoniazid metabolism[30].Subsequent investigations of halothane,antipyrine,and phenytoin metabolism in twins revealed the major influence of genetic factors and exposure to disease-favoring environment[31,32].
Influence of polymorphisms in genes encoding phase-I drug metabolizing enzymes
Cytochrome P450 2D6
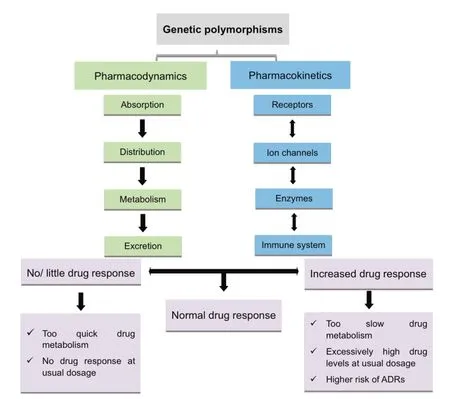
Figure 1 Effect of genetic polymorphisms on individuals’drug responsePharmacokinetics and pharmacodynamics are main determinants of interindividual differences in drug responses.Genetic polymorphism in genes related to these processes may result in mild to severe variations in drug responses.ADRs,adverse drug reactions.
Cytochrome P450(CYP),which represents a large and diverse group of heme-containing enzyme superfamily,is involved in oxidative metabolism of structurally-diverse molecules likedrugs,chemical,and fatty acids.The genetic polymorphism in the genes encoding CYP members was firstly reported for CYP2D6.The highly polymorphic CYP2D6 gene is located on the chromosome 22q13.1,consisting of nine exons and eight introns(GenBank accession No.NM 000106.5)[33,34]. More than 100 CYP2D6 genetic variants have been described(http://www.cypalleles.ki.se/cyp2d6.htm)todate,resulting from point mutations,duplication,insertions or deletions of single or multiple nucleotides,and even whole-gene deletion. Individuals carrying different CYP2D6 allelic variants have been classified as poor metabolizers(PMs),intermediate metabolizers(IMs),extensivemetabolizers(EMs),and ultrarapid metabolizers(UMs)according to the metabolic nature of the drugs and degree of involvement in drug metabolism of these variants[35].Although constituting only 2%-4%of the total amount of CYPs in the liver,CYP2D6 actively metabolizes approximately 20%-25%of all administered drugs[36].The drugs metabolized by CYP2D6 include tricyclic antidepressants,serotonin reuptake inhibitors,antiarrhythmics,neuroleptics,and β-blockers[35].
The extensive presence of polymorphism in the CYP2D6 gene significantly affects phenotypic drug responses.Up to a 10-fold difference in the required dose was observed in order to achieve the same plasma concentration in different individuals[37].Dextromethorphan,debrisoquine,bufuralol and sparteine are the probe drugs used for in vivo CYP2D6 phenotyping.According to the probe substrate metabolic capabilities among the sampled individuals in a population,patients can be categorized into the following four phenotypic groups:poor,intermediate,extensive,and ultra-rapid metabolizers(PMs,IMs,EMs,and UMs),respectively[38].The interindividual phenotypic variations depend on the metabolic properties of the CYP2D6 allelic variants(Table 1).Simultaneous presence of two null(non-functional)alleles in an individual[39]confers a PM phenotype,whereas individuals with two normally-functioning alleles[40]present with the EM phenotype.In addition,co-existence of a null allele with another allele associated with reduced function[41,42]gives rise to an IM phenotype,whereas presence of extra CYP2D6 gene copies with normal activity confers the UM phenotype. According to the CYP2D6 phenotype,the Caucasian population comprises approximately 5%-10%PMs,10%-17%IMs,70%-80%EMs,and 3%-5%UMs[39].The percentages of PMs,IMs,EMs,and UMs differs among different ethnicities due to the significant variability in the CYP2D6 allele distribution(Table S1 and Table S2).
Individuals with the UM phenotype can metabolize the administered CYP2D6 substrates in much shorter time than individuals with the IM or PM phenotypes[43].This leads to very low plasma drug levels with potential loss of drug efficacy.Therefore,higher drug doses would be required to attain effective drug concentrations,which could be fatal when dealing with drugs with narrow therapeutic indexes.Notably,a large number(approximately 10%-30%)of Saudi Arabians and Ethiopians have been reported to have the CYP2D6*2XN allele[44,45].On the other hand,there is an opposite situation for the individuals with the CYP2D6*3,*4,*5,and*6 alleles(PM phenotype).These allelic variants lead to inactive CYP2D6 enzymes[46-50].As a result,the affected individuals exhibit high plasma drug levels with increased risks of drugrelated side effects and therefore reduced drug dose should be administered[51].The allelic frequencies with clinical consequences of CYP2D6*3(3.3%in Sardinians),CYP2D6*4(23%-33%in Polish and Faroese populations),CYP2D6*5(5.9%-6.2%in Spaniards and African Americans),and CYP2D6*6(1.9%-3.3%in Faroese and Italians)were also calculated in diverse populations(Table S2).
The prodrug tamoxifen is a selective estrogen receptor(ER)modulator used to treat ER-positive breast cancer patients[52].Tamoxifen is actively catalyzed to endoxifen and 4-hydroxytamoxifen by various CYPs with CYP2D6 acting as the rate-limiting enzyme[53].Plasma level of endoxifen in UM patients is usually higher than that in PM and IM patients due to the presence of multiple functional CYP2D6 copies[53]. The presence of CYP2D6 null alleles in high frequencies commonly contributes to the CYP2D6 PM phenotype in individuals,as is the case with the CYP2D6*4(33%)in the Faroese population[47].In tamoxifen-treated surgically resected ER-positive breast cancer patients,a much lower(0)prevalence of moderate to severe hot flashes,together with a higher risk ofdiseaserelapse,wasreportedinwomenwiththe CYP2D6*4/*4 genotype than in patients with one or no CYP2D6*4 alleles(20%)[54].Codeine is a commonly prescribed analgesic,which is converted to its active metabolite morphine and acts at mu-opioid receptors to induce analgesia. The affinity of morphine to mu-opioid receptors is 200-fold stronger than that of codeine[55].Interestingly,conversion from codeine to morphine is also catalyzed by CYP2D6,which has been proven as the key enzyme responsible for the analgesic effect of codeine.The CYP2D6 phenotype is therefore a critical determinant in opioid analgesia.According to McLellan et al.[45],subjects with the PM phenotype can only convert 10%of a codeine dose to morphine while approximately 40%and 51%conversion occurs in EMs and UMs,respectively.Thus,in individuals with null allelic variants of CYP2D6,codeine is not recommended as an analgesic because of the minimal enzymatic conversion from codeine to morphine.Conversely,a higher risk of morphine toxicity may occur in patients with the UM phenotype owing to the rapid conversion of codeine to morphine.The situation would bemore devastating in UMs who are lactating mothers because the normal codeine dose can translate into fatal morphine concentrations into the breast milk[56].In 2006,a case of a 13-day newborn death was reported when the infant's mother was placed on the codeine therapy after delivery for pain management of episiotomy[57].There are also other cases reporting that the routinely recommended codeine doses produced lethal adverse effects in UM patients[56,58,59].The CYP2D6 allelic variants*10,*17,and*41 exhibit normal catalytic activity but are sometimes associated with intermediate to low metabolic activities[60].In the Chinese population,the CYP2D6*10 allele has been found more common than other alleles(allelic frequency of up to 65%)and it causes a greatly decreased(but not deficient)enzyme activity[61].

Table 1 CYP2D6 genotype-based phenotype groups of individuals
CYP2C9
CYP2C9 is another important member of the CYP superfamily.The gene coding for CYP2C9 is located on chromosome 10q24.2,and spans more than 55 kb in length.CYP2C9 constitutes approximately 18%of the total CYP protein in the human liver microsomes[62].CYP2C9 metabolizes approximately 25%of clinically-administered drugs including antiinflammatory agents such as flurbiprofen,hypoglycemic agents such as glipizide and tolbutamide,the anticoagulant S-warfarin,and the anticonvulsant phenytoin[63,64].More than 60 variant alleles have been identified for the CYP2C9 gene(http://www.cypalleles.ki.se/cyp2c9.htm).Amongthem,CYP2C9*2(R144C)and CYP2C9*3(I359L)are the most common variants associated with highly-reduced CYP2C9 enzymatic activities in comparison with the wild-type allele(CYP2C9*1)[65].
The CYP2C9*2 variant results in a markedly decreased enzyme activity due to higher km value and lower intrinsic clearance of drugs like S-warfarin[16].The CYP2C9*2 allelic variant has been reported with up to 25%allelic frequencies in the Iranian population[66].However,frequencies of heterozygous CYP2C9*1/*2,homozygous CYP2C9*2 or CYP2C9*3 carriers were lower(0.1%-1%)in the Chinese and Japanese populations compared with those in Caucasians and Iranians. Caucasians have approximately 1%CYP2C9*2 and 0.4% CYP2C9*3homozygotes,respectively [67].Furthermore,approximately one-third of the Turkish population has either the*1*2 or the*1*3 genotype,while more than 2%have the *2*2,*2*3,and*3*3 genotypes[68].In the Iranian and Pakistanipopulations,theprevalenceofCYP2C9*2and CYP2C9*3 is greater than that in the other studied populations[66].On the other hand,Chinese,Vietnamese,Korean,Bolivian,and Malaysian populations have a CYP2C9*1 allelic frequency variant of>90%,whereas allelic CYP2C9*2 variant was not detected in the Korean,Chinese,and Vietnamese populations but occurs 1%in the Japanese.Furthermore,no individuals from the South African and Zimbabwean populations have been reported to carry the CYP2C9*2 allele(Table S1).
Theinterindividualandinterethnicvariationsinthe CYP2C9 polymorphisms are clinically significant especially in the patients on anticoagulation therapy with warfarin. Warfarin is one of the most widely-prescribed oral anticoagulants[13].Clinically-available warfarin is a racemic mixture of the R and S enantiomers,with the S-isomer exhibiting an approximately 5-fold higher anticoagulant potency than the R-isomer[69].Inactivation of the active S-warfarin is almost exclusively mediated by CYP2C9.Patients with high allele frequencies of the CYP2C9 wild-type or CYP2C9*1 excrete the S-warfarin normally from the body.In contrast,PMs who have high allelic frequencies of the CYP2C9*2,CYP2C9*3,or both have impaired S-warfarin-metabolizing capabilities and,therefore,require lower drug doses to attain therapeutic responses[70-73].Thus,PMs have higher risks of internal bleeding than individuals with higher CYP2C9*1 allelic frequencies during warfarin therapy[69,72].Although polymorphisms in genes encoding blood-clotting factors also contribute to the bleeding risk and initial warfarin dose adjustment requirements,CYP2C9 gene polymorphisms always exert greater influence[74].
Both CYP2C9 and CYP2C19 are involved in microsomal hydroxylation of phenytoin to its R and S enantiomers[75]. Therefore,CYP2C9 genotype is an important determinant in in vivo phenytoin metabolic studies.Due to the narrow therapeutic range of phenytoin,even minimal variations in CYP2C9 activity can be clinically important[76].In a study on healthy Turkish individuals with already known CYP2C9 genotypes,Aynacioglu et al.[68]reported that subjects with CYP2C9*1/*2,CYP2C9*1/*3,and CYP2C9*2/*2 genotypes had significantly higher phenytoin serum concentrations and lowerlevelsof5-(4-hydroxyphenyl)-5-phenylhydantoin(phenytoin metabolite)than those with the CYP2C9*1/*1 genotype.Multiplestudieshavealsoshownthatthe CYP2C9*3/*3 genotype is associated with reduced metabolisms and altered pharmacokinetic properties of substrates such as phenytoin,warfarin,losartan,and tolbutamide[77-80].
CYP2C19
The polymorphic CYP2C19,which is located on the chromosome 10q24 encodes another CYP family member.CYP2C19 can metabolize numerous routinely-administered drugs such as anxiolytics(diazepam),proton pump inhibitors(omeprazole),anticonvulsants(S-mephenytoin),andantimalarial biguanides[35,81-83].Up to now,more than 35 CYP2C19 variants and approximately 2000 SNPs have been identified(http://www.cypalleles.ki.se/cyp2c19.htm),withcontinuous increase in SNP numbers reported.Among them,CYP2C19*2 and CYP2C19*3 are the most common variants that have been studied extensively.Both of them are null variants and patients carrying these variants are therefore categorized as PMs. CYP2C19*2 is the most common allelic variant caused by a single nucleotide alteration in exon 5(G>A),resulting in an abnormal splicing site and conferring reduced enzymatic activities of CYP2C19[83,84].
The CYP2C19*2 variant is found at a high allelic frequency(30%)in South Indians,but occurs with the lowest frequency(2.9%)in the Faroeses.In contrast,CYP2C19*3 is found at higher allelic frequencies in the Japanese(approximately 13%)but lower(0)among the Italians,South Africans,Greeks,European-Americans,and other populations(Tables S1 and S2).Approximately 15%-25%of the Korean,Japanese,and Chinese populations have been reported as PMs of the anticonvulsant drug S-mephenytoin[85-87].The activity of omeprazole,a drug recommended for treating peptic ulcers and gastroesophageal reflux diseases,was found to be highlypatient CYP2C19 genotypes dependent[88].Furuta et al.[89]found that after a single dose(20 mg)of omeprazole[90],the observed intragastric pH values were 4.5,3.3,and 2.1 for PMs,heterozygous EMs,and EMs individuals,respectively. In another study,Schwab et al.[91]reported lower serum concentrations of lansoprazole,a proton pump inhibitor,and lower rates of Helicobacter pylori eradication in Caucasian EM patients following a standard dose of lansoprazole.The individuals with the PM phenotype of CYP2C19 required lower doses of the proton pump inhibitor lansoprazole for beneficial therapy than that required by the patients with the EM phenotype of CYP2C19[92].Both CYP2C19*2 and CYP2C19*3 variant alleles of CYP2C19 are associated with inactive enzyme production,which is evident from the various population studies summarized in Table S1.Some drugs strongly affected by CYP2C19 genotypes,and their labels contain pharmacogenomic information are summarized in Table 2.
CYP3A4 and CYP3A5
More than 50%of clinically-administered drugs are metabolized by CYP3A4,which is the most abundant CYP enzyme in the liver[93].Therefore,polymorphisms in CYP3A4 are of great concern in the study of interindividual altered drug metabolisms and related ADRs[94].More than 26 CYP3A4 variantshavebeenidentified(http://www.cypalleles.ki. se/cyp3a4.htm)and most of these variants are responsible for varied enzyme activities ranging from modest to highly reduced catalytic efficiencies among the affected individuals[35].Comparatively,high frequencies of allelic variants of the CYP3A4 gene(CYP3A4*2 and CYP3A4*3)were observed in Caucasian whereas high frequencies of allelic variant CYP3A4**18 were observed in Chinese people(Table S1). The clinical consequences of different allelic variants of CYP3A4 are still undefined for many substrates of CYP3A4. Considering the relatively low frequencies,only small changes in the enzyme activity have been caused by CYP3A4*16 and CYP3A4*18 variants[95].
CYP3A5 is one of the factors that contribute to the complexity of CYP3A4.With few exceptions,CYP3A5 can metabolize most drugs that are substrates of and metabolized by CYP3A4.Although slower in most cases[96],the metabolic activity of CYP3A5 is equal[97]to or even faster than that of CYP3A4 in some cases[98].In vivo studies revealed that the metabolic rates for the drug that are metabolized by both CYP3A4 and CYP3A5 are the sum of the activities of both enzymes.FunctionallyactivevariantsofCYP3A5are expressed in half of the African population and one-fourth of Caucasians[99].This may partially explain why human studies of the CYP3A4 allelic variants do not agree with its clinical effects[100].An overview of the important consequences of gene mutations of the CYPs is illustrated in Figure 2.
CYP oxidoreductase(CYPOR)is the catalytic partner and compulsory element to all CYP-mediated metabolisms.The interaction between the CYP and CYPOR is essential for the metabolic activities of CYPs[14].CYPOR is required for electron transfer from NADPH to CYP via its FAD and FMN domains,whichiscrucialforCYPcatalyticactivities[101,102].Therefore,CYPOR allele variants like POR*5, POR*13 and POR*27 can indirectly alter the functional consequences of CYPs[103-105].For example,in POR*27 variant,L577P mutation located in the NADPH-binding domain of CYPOR [102]leads to decreased CYPOR activity,due to changed helix and disrupted NADPH interaction[105],whereas POR*5(A287P)is associated with impaired ability toacceptelectronsfromNADPH[106].Additionally,POR*13(Q153R)variant leads to severely-impaired steroid biosynthesis in Antley-Bixler skeletal malformation syndrome(ABS)[107].Until now,more than 50 different variants of the human CYPOR genes have been described(http://www. cypalleles.ki.se/por.htm).
Effect of polymorphisms in genes encoding drug transporters
A drug could produce a beneficial or toxic effect in a particular patient.The nature and extent of the resulting effect is largely dependent on the absorption,distribution,and excretion rates of the drug.Drug transporters primarily control the movement of all drugs and their active or inactive metabolites into or out of cells.Therefore,polymorphisms of drug transporter genes can modify the absorption,distribution,and excretion rates,and ultimately safety and efficacy of the administered drugs. The ABC and solute-carrier(SLC)transporters are two superfamilies of transport proteins are ubiquitous membrane-bound transport proteins that are involved in the absorption,distribution,and elimination of drugs[92].
ABC transporters often transport drugs and other substances against the concentration gradient using ATP as an energy source[108].In ABC transporter superfamily of drug transporters,49 genes have been identified,which are divided into seven subfamilies from ABCA to ABCG(http://nutrigene.4t.com/humanabc.htm).The impact of some important polymorphisms on the drug transport activities of various ABC transporters is summarized in Figure 3.In addition,approximately 360 genes have been identified in the SLC superfamily and are classified into 46 subfamilies(http:// www.bioparadigms.org/slc/menu.asp).Among them,members of the organic anion transporter(OAT),organic anion transporting polypeptides(OATP),and organic cation transporter(OCT)subfamilies are of particular significance in drug disposition[109].In addition,polymorphisms in genes encoding SLCO,SLC22,and SLC47 family members within the SLC superfamily have key roles in modulating drug transport activities of the corresponding transporters(Figure 4).
ABCB1
The ABCB1 gene,also known as the multidrug resistance 1(MDR1),encodes a P-glycoprotein(Pgp),which is involved in the cellular efflux of numerous chemotherapeutic agents,physiological metabolites,and carcinogens[110].ABCB1 is highly polymorphic with allelic variants found in varied frequencies in different populations(Table S3).ABCB1 polymorphisms were identified firstly by Kioka et al.[111]in different cancer cell lines in 1989 and subsequently by Hoffmeyer et al.[112]and other researchers[113-116].As an efflux transporter,ABCB1 is detected on the surface of epithelial cells,preventing intestinal absorption,protecting fetus and brain from xenobi-otic exposure and facilitating renal and hepatobiliary excretions[117].Interestingly,overexpression of the ABCB1 gene in cancer cells induced resistance to chemotherapeutic agents[110].

Table 2 CYP2C19 genetic polymorphisms with their clinical consequences
Distribution of some allelic variants appears to be ethnicitydependent.For instance,SNP 3435C>T occurs at high frequencies(60%-72%)in Asians but low(34%-42%)in Caucasians.Thesubstrate-dependenteffectsofPgpon pharmacokinetic and pharmacodynamics properties remain obscure due to controversial studies on digoxin disposition. For example,for patients with a mutant allele(3435C>T)that were administered a single oral dose of digoxin,Sakaeda et al.[118]reported lower serum concentrations of digoxin,whereas higher plasma digoxin levels were observed by Verstuyftandhercolleagues[119].Thehaplotype 1236C>T/2677G>T/3435C>T was detected with high frequency(up to 56%)in Asians[120].Kimchi-Sarfaty et al.[121]found that patients carrying this haplotype exhibited normal transporter properties although the transporter inhibition by small modulators was affected.The conflicting results of these studies could be indicative of additional polymorphisms yet-to-be-identified other than the studied mutations or might reflect the complex disposition pathways of the substrate drugs in the studied subjects.For example,cyclosporine,a CYP3A4 substrate that is a widely-used immunosuppressant in patients with liver,kidney,or heart transplants,is also transported by ABCB1[122].Similarly,fexofenadine and digoxin can be simultaneously transported by OATP and ABCB1.Letourneau et al.[123]studied the transport activity of ABCB1 with R230Q,R633Q,R1056Q,R723Q,T73I,S1512L,S92F,T117M,A989T,or C1047S nonsynonymous SNPs by using different substrates(methotrexate,leukotriene C-4,and estra diol-17-β-glucuronide).However,they failed to find any significant effect of the aforementioned variants on either gene expression levels or transport functions.Conversely,a 50% reduction in transport activity was observed in the A989T variant [124].ComparedtoAsiansandCaucasians,the 3435C>T allele occurs lowly in Africans,and it has been proposed that this low frequency of the MDR1 3435T allele might be associated with the reduced incidence of renal carcinoma in African populations[124].On the other hand,the MDR1 3435C allele might have a protective role in parkinsonism patients with a known history of pesticide exposure[125].
ABCC1 and ABCC2
As the important ABC members,both ABCC1 and ABCC2 are involved in the transport and excretion of several chemotherapeuticagents,toxicants,andorganicanion molecules[128].Glutathione cotransporter is essential for both of them to transport some substrates such as estrone sulfate[126].In non-Hodgkin lymphoma patients treated withdoxorubicin,significantassociationsbetweenthe G671V variant and a V188E-C1515Y haplotype of ABCC2 and G671V variant with 28%allelic frequency in Caucasians have been reported[127,128].V417I is another widely distributed variant in ABCC2(Asians 13%-19%,Africans 14%,and Caucasians 22%-26%)that has been extensively studied for its role in drug resistance development in cancer and human immunodeficiency virus type 1(HIV-1)-infected patients[129-131].
ABCG2
SimilartoABCC1,ABCG2wasfirstdiscoveredin multidrug-resistant cell lines[132],which is also known as the breast cancer resistance protein(BCRP),mitoxantrone resistance protein(MXR)or placenta-specific ABC protein(ABCP)[133].ABCG2,which is expressed in the epithelial cells of the small intestine,lung,kidney,sweat glands,colon,and placenta,is essential for intestinal absorption and biliary excretion of drugs and their metabolites and xenobiotic[134].More than 80 polymorphisms of the ABCG2 gene have been identified[135].Among them,SNP C421A in the variant(p.Q141K)has been found to be associated with thereducedexpressionandalteredsubstratespecificity ABCG2[136].
The C421A is widely distributed in many ethnicities with frequencies of 27%-35%in Asians,9%-14%in Caucasians,and 1%-5%in Africans(Table S3).Gefitinib,the inhibitors of epidermal growth factor receptor(EGFR)tyrosine kinase,are substrates of ABCG2.In cancer patients who were treatedwithgefitinib,presenceofC421Awasrelatedto increaseddrugaccumulationandhigherprevalenceof drug-induced grade 1 or 2 diarrhea[137,138],when compared to patients with wild type allele.In another study,Sparreboom et al.[139]reported a 300%elevation in plasma levels of the anticancer drug diflomotecan in individuals with theheterozygousC421Agenotypewhenthedrugwas administered intravenously[139].Presence of C421A also affects the pharmacokinetic and therapeutic effects of rosuvastatin in Chinese and Caucasians.Tomlinson et al.[16]reported the significant influence of C421A in reducing LDL cholesterol levels in a gene-and dose-dependent mannerinChinesepatientswithhypercholesterolemia[16]. Therefore,a systemic analysis of polymorphisms of ABC transporters would be essential to enhance the understanding of the genetic impact on pharmacotherapy.

Figure 2 An overview of important consequences of genetic polymorphisms in the CYPsOverview of the effect of genetic polymorphisms on some human cytochrome P450 variant alleles and molecular mechanisms leading to altered drug metabolism.
OATPs
OATPs are a large family of membrane-bound influx transporters that are responsible for the cellular uptake of a wide range of endogenous and exogenous substances including bile salts,hormones,and clinically administered drugs such as antibiotics,cardiac glycosides,and anticancer agents[140]. There are 11 human OATP transporters,among which OATP1A2,OATP1B1,OATP1B3,OATP2B1,and OATPC are involved in drug pharmacokinetics[138].In particular,the OATPC*5 and OATPC*9 allelic variants are associated with a reduced uptake of OATPC substrates such as estrone sulfate and estradiol-17-β-D-glucuronide[141].High plasma levels of pravastatin and repaglinide have been reported in subjects carrying the OATPC*5 allele[140-143].
On the other hand,OATP1B1,OATP2B1,and OATP1B3 are mainly expressed on the hepatocyte sinusoidal membrane,which can facilitate the hepatic drug uptake[138].OATP1B1 is encoded by SLCO1B1 and is essential for the hepatic uptake of the simvastatin active metabolite,simvastatin acid[144].Six important SNPs identified in the SLCO1B1 gene with their allelic frequencies and functional consequences in Asian,African and Caucasian have been discussed in Table S3.Among them,the 521T>C variant of the SLCO1B1 is associated with reduced OATP1B1 activity,which is responsible for the higher blood concentrations of simvastatin acid,as well as the consequently increased toxicity and reduced efficacy of simvastatin[145].In addition,OATP1B1*15 was associated with increased plasma concentrations of pravastatin and 7-ethyl-10-hydroxycamptothecin(irinotecan active metabolite),whereas OATP1B1*17 variant is linked with an increased cholesterol synthesis mediated by pravastatin[146-148].
OCTs

Figure 3 The influence of genetic polymorphisms of ABC transporters on the drug transport activitiesThe diagram depicts the influence of genetic polymorphisms on the drug transport activities of ABC transporters.ABC transporter,ATP-binding cassette transporter;MDR1,multidrug resistance protein 1;BCRP,breast cancer resistance protein;MRP,multidrug resistance-associated protein.
OCTs are proteins encoded by the SLC22A family and in humans,which are present in the basolateral cell membrane of the renal proximal tubule[149].Three isoforms,OCT1,OCT2,and OCT3,have been identified in humans[150-152]and OCT2 is highly expressed in the kidneys.OCTs mediate the cellular uptake of a wide range of structurally-different organic cations including clinically-administered drugs such as metformin and procainamide[150].Metformin,a therapeutic agent used to treat type 2 diabetes mellitus,is predominantly renally excreted[153].The OCT2 270S variant has been associated with low activity while the 270A variant induces high activity of OCT2[153,154].Patients with type 2 diabetes who are homozygous for the 270A variant exhibit a significantly higher renal clearance and lower plasma concentration of metformin than those with the homozygous 270S variant[153-155].On the other hand,allele variants G401S,R61C,G465R,and M420del are associated with lower OCT1 activities,which are responsible for the significantly increased renal clearance and reduced glucose-lowering effects of metformin in healthy subjects[156].
Influence of genetic polymorphisms of drug metabolizing enzymes or transporters on drug-drug interactions
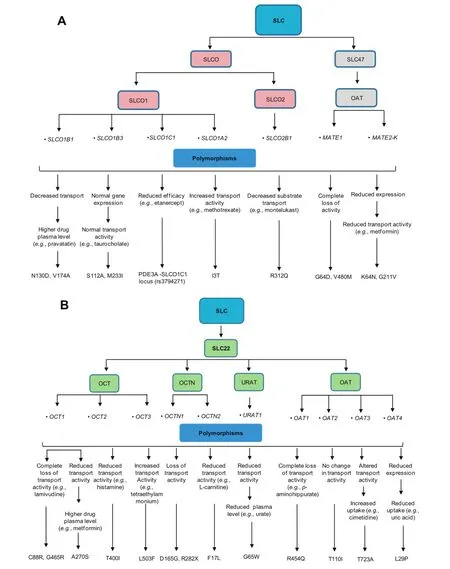
Figure 4 Modification of drug transport activities of SLC transporters by genetic polymorphismsThe diagram depicts the influence of genetic polymorphisms on the transport activities of different allele variants of SLC transporters including SLCO and SLC47(A)as well as SLC22(B).SLC,solute carrier;SLCO,solute carrier organic anion;OCT,organic cation transporter;OCTN,organic cation transporter novel;OAT,organic anion transporter;MATE1,multidrug and toxin extrusion protein 1;URAT,urate transporter.
Effects of one drug are modified by other concomitantly administered drugs due to drug-drug interactions,which may be attributed to the altered pharmacokinetic or pharmacodynamic properties of one drug induced by the coadministered drug.The polymorphisms in drug metabolizing and transporter genes are an important risk factor of drug-drug interactions and varied interindividual drug responses[157]. These polymorphisms can lead to decreased levels of a drugmetabolizing enzyme in an individual,which may cause severe adverse drug reactions following the coadministration of enzyme inhibitors[158,159].Among the CYPs,CYP2C9,CYP2C19,and CYP2D6 are involved in the metabolism of approximately 40%of routinely administered drugs[160]. Different CYP allelic variants significantly contribute to the variability of an individual's susceptibility to drug-drug interactions and drug-metabolizing capacities[161].Different drugs interact with the CYP metabolic machinery differently. The metabolism of some drugs by CYP enzymes is extremely specific,for example,metoprolol is primarily metabolized by CYP2D6[162],whereas other drugs such as warfarin may be simultaneouslymetabolizedbyseveralCYPsincluding CYP2D6,CYP3A4,and CYP1A2[163].Polymorphisms related to the altered expression of drug metabolizing and transporter genes will ultimately affect the therapeutic effectsof administered drugs[73,164].When a drug is metabolized by more than one CYP metabolic pathway and the administered drug acts by inhibiting or inducing CYPs,genetic polymorphisms could redirect the metabolism of drugs via other CYP routes[162].This could lead to drug-drug interactions. For example,antifungal voriconazole is actively metabolized by CYP3A4 and CYP2C19,whereas ritonavir strongly inhibits CYP3A4whileinducingCYP2C19metabolicactivities[165,166].When CYP2C19 PM patients are treated with voriconazole and ritonavir,up to 461%increased AUC of voriconazole was observed,since the patients were unable to metabolize voriconazole owing to reduced CYP2C19 and CYP3A4 activities[167,168].In another case,the antiplatelet activity of clopidogrel was reduced when it was administered with proton pump inhibitors such as esomeprazole and omeprazole owing to the inhibition of CYP2C19 [169],whereas an increased activity of clopidogrel was anticipated in the presence of rifampicin and aspirin[170].Clopidogrel is a prodrug that needs oxidative activation in vivo by CYP1A2,CYP2B6 and CYP2C19 for its anti-platelet activity[171]. Genetic polymorphisms in CYP2C19,CYP1A2,2B6*6,and CYP3A5*3 were found to be associated with the varied degree of drug-drug interactions for clopidogrel,due to its highlycomplex pharmacokinetics and variable drug response as compare to other anti-platelet drugs[172-176].
Mutations in the drug transporter genes also contribute to drug-drug interactions and adverse drug reactions.HMGCR inhibitors such as atorvastatin,rosuvastatin,and pravastatin are actively transported by OATP1B1 and ABCG2[147]. The concomitant administration of cyclosporine(a potent inhibitor of OATP1B1 and ABCG2)with statins like rosuvastatin and pitavastatin will result in higher plasma levels of statins,leading to rhabdomyolysis[177].Digoxin is potently cleared by MDR1,therefore its coadministration with verapamil,clarithromycin,or talinolol that inhibits MDR1 transportactivityleadstoincreasedplasmalevelsdueto decreased renal clearance of the drug[178,179].
Conclusions
The genetic variations of CYPs and transporters have been described in diverse populations.In this review,we review the different allelic variants that are responsible for altered drug activities in diverse geographic regions.Some populations exhibited extremely high frequencies of allele variants that are associatedwithseveralsignificantclinicalconsequences. Taking advantage of pharmacogenomics,researchers have assessed some specific genetic variants responsible for the particular drug responses of individuals.
WholegenomeSNPprofiling,haplotyping,multigeneanalysis,andgeneexpressionstudiesbybiochipormicroarraysareall in place to study drug responses of individuals,which would aid in drug discovery,development,and individualized treatments. Given the common variability in drug responses among patients,the optimization of dosage regimen at the individual levelisnotaneasytask.Comprehensiveappreciationofthecontributing factors associated with interindividual and interethnic differences in medication responses is a must for the development of precision medicine,and help health-care professionals in recommending the proper treatment with the best possible beneficial outcomes while preventing unwanted drug effects in the particular patients.The development of clinical practice strategies based on accurate genotype testing will facilitate the enhanced understanding of altered drug responses and drugdrug interactions.Furthermore,the development of more reliable biomarkers based on polymorphisms in genes responsible for the adverse events will hopefully create strategies for administering drugs based on the genotype and phenotype of patients,to minimize unwanted drug reactions.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the Major National R&D Projects(Grant No.2012ZX09506001-004)and National Natural Science Foundation of China(Grant No.81273578).
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2016.03.008.
References
[1]Collins FS.Of needles and haystacks:finding human disease genes by positional cloning.Clin Res 1991;39:615-23.
[2]Roden DM,George Jr AL.The genetic basis of variability in drug responses.Nat Rev Drug Discov 2002;1:37-44.
[3]Sachidanandam R,Weissman D,Schmidt SC,Kakol JM,Stein LD,Marth G,et al.A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms.Nature 2001;409:928-33.
[4]Pirmohamed M.Genetic factors in the predisposition to druginduced hypersensitivity reactions.AAPS J 2006;8:E20-6.
[5]Meyer UA,Zanger UM,Schwab M.Omics and drug response. Annu Rev Pharmacol Toxicol 2013;53:475-502.
[6]Ma Q,Lu AY.Pharmacogenetics,pharmacogenomics,and individualized medicine.Pharmacol Rev 2011;63:437-59.
[7]Roden D,Tyndale R.Genomic medicine,precision medicine,personalized medicine:what's in a name?Clin Pharmacol Ther 2013;94:169-72.
[8]Dolsten M,Søgaard M.Precision medicine:an approach to R&D for delivering superior medicines to patients.Clin Transl Med 2012;1:7.
[9]Marchant B.Pharmacokinetic factors influencing variability in human drug response.Scand J Rheumatol Suppl 1981;10:5-14.[10]Wilkinson GR.Drug metabolism and variability among patients in drug response.N Engl J Med 2005;352:2211-21.
[11]Spear BB,Heath-Chiozzi M,Huff J.Clinical application of pharmacogenetics.Trends Mol Med 2001;7:201-4.
[12]Evans WE,McLeod HL.Pharmacogenomics—drug disposition,drug targets,and side effects.N Engl J Med 2003;348:538-49.
[13]Rettie AE,Tai G.The pharmocogenomics of warfarin:closing in on personalized medicine.Mol Interv 2006;6:223-7.
[14]Chasman DI,Posada D,Subrahmanyan L,Cook NR,Stanton Jr VP,Ridker PM.Pharmacogenetic study of statin therapy and cholesterol reduction.JAMA 2004;291:2821-7.
[15]Davidson MH,Stein EA,Dujovne CA,Hunninghake DB,Weiss SR,Knopp RH,et al.The efficacy and six-week tolerability of simvastatin 80 and 160 mg/day.Am J Cardiol 1997;79:38-42.
[16]Tomlinson B,Hu M,Lee V,Lui S,Chu T,Poon E,et al.ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin.Clin Pharmacol Ther 2010;87:558-62.
[17]Akhondzadeh S.Personalized medicine:a tailor made medicine. Avicenna J Med Biotechnol 2014;6:191.
[18]Kalow W,Tang BK,Endrenyi L.Hypothesis:comparisons of inter-and intra-individual variations can substitute for twin studies in drug research.Pharmacogenetics 1998;8:283-9.
[19]Vesell ES.Pharmacogenetic perspectives gained from twin and family studies.Pharmacol Ther 1989;41:535-52.
[20]Akhondzadeh S,Jafari S,Raisi F,Nasehi AA,Ghoreishi A,Salehi B,et al.Clinical trial of adjunctive celecoxib treatment in patients with major depression:a double blind and placebo controlled trial.Depress Anxiety 2009;26:607-11.
[21]Ferrara J.Personalized medicine:challenging pharmaceutical anddiagnosticcompanybusinessmodels.McgillJMed 2007;10:59-61.
[22]Borges JB,Hirata TD,Cerda A,Fajardo CM,Cesar RC,Franc¸a JI,et al.Polymorphisms in genes encoding metalloproteinase 9 and lymphotoxin-alpha can influence warfarin treatment.J Pharmacogenomics Pharmacoproteomics 2015;6:1.
[23]Siest G.The european society of pharmacogenomics and personalised therapy-ESPT.J Pharmacogenomics Pharmacoproteomics 2015;6:144.
[24]Nair SR.Personalized medicine:striding from genes to medicines.Perspect Clinical Res 2010;1:146-50.
[25]Pirmohamed M.Personalized pharmacogenomics:predicting efficacy and adverse drug reactions.Annu Rev Genomics Hum Genet 2014;15:349-70.
[26]Pirmohamed M,Park BK.Genetic susceptibility to adverse drug reactions.Trends Pharmacol Sci 2001;22:298-305.
[27]Lin JH.Pharmacokinetic and pharmacodynamic variability:a dauntingchallengeindrugtherapy.CurrDrugMetab 2007;8:109-36.
[28]Eichelbaum M,Ingelman-Sundberg M,Evans WE.Pharmacogenomics and individualized drug therapy.Annu Rev Med 2006;57:119-37.
[29]Dietlein F,Thelen L,Reinhardt HC.Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches.Trends Genet 2014;30:326-39.
[30]Evans DA,Manley KA,McKusick VA.Genetic control of isoniazid metabolism in man.Br Med J 1960;2:485-91.
[31]Vessell ES,Penno MB.A new polymorphism of hepatic drug oxidation in man:family studies on rates of formation of antipyrine metabolites.Biochem Soc Trans 1984;12:74-8.
[32]Nebert DW,Zhang G,Vesell ES.Genetic risk prediction:individualized variability in susceptibility to toxicants.Annu Rev Pharmacol Toxicol 2013;53:355-75.
[33]Kimura S,Umeno M,Skoda RC,Meyer UA,Gonzalez FJ.The human debrisoquine 4-hydroxylase(CYP2D)locus:sequence and identification of the polymorphic CYP2D6 gene,a related gene,and a pseudogene.Am J Hum Genet 1989;45:889-904.
[34]Gough AC,Smith CA,Howell SM,Wolf CR,Bryant SP,Spurr NK.Localization of the CYP2D gene locus to human chromosome 22q13.1 by polymerase chain reaction,in situ hybridization,and linkage analysis.Genomics 1993;15:430-2.
[35]Zhou SF,Di YM,Chan E,Du YM,Chow VD,Xue CC,et al. Clinical pharmacogenetics and potential application in personalized medicine.Curr Drug Metab 2008;9:738-84.
[36]Ingelman-Sundberg M.Genetic polymorphisms of cytochrome P4502D6 (CYP2D6):clinicalconsequences,evolutionary aspectsandfunctionaldiversity.PharmacogenomicsJ 2005;5:6-13.
[37]Kirchheiner J,Nickchen K,Bauer M,Wong M,Licinio J,Roots I,et al.Pharmacogenetics of antidepressants and antipsychotics:the contribution of allelic variations to the phenotype of drug response.Mol Psychiatry 2004;9:442-73.
[38]Zanger UM,Raimundo S,Eichelbaum M.Cytochrome P450 2D6:overview and update on pharmacology,genetics,biochemistry.Naunyn Schmiedebergs Arch Pharmacol 2004;369:23-37.
[39]Sachse C,Brockmo¨ller J,Bauer S,Roots I.Cytochrome P450 2D6 variants in a Caucasian population:allele frequencies and phenotypic consequences.Am J Hum Genet 1997;60:284-95.
[40]Marez D,Legrand M,Sabbagh N,Lo Guidice JM,Spire C,Lafitte JJ,et al.Polymorphism of the cytochrome P450 CYP2D6 gene in a European population:characterization of 48 mutations and 53 alleles,their frequencies and evolution.Pharmacogenetics 1997;7:193-202.
[41]Bradford LD.CYP2D6 allele frequency in European Caucasians,Asians,Africans and their descendants.Pharmacogenomics 2002;3:229-43.
[42]Ji L,Pan S,Marti-Jaun J,Ha¨nseler E,Rentsch K,Hersberger M. Single-step assays to analyze CYP2D6 gene polymorphisms in Asians:allele frequencies and a novel*14B allele in mainland Chinese.Clin Chem 2002;48:983-8.
[43]Borges S,Desta Z,Li L,Skaar TC,Ward BA,Nguyen A,et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism:implication for optimization of breast cancer treatment.Clin Pharmacol Ther 2006;80:61-74.
[44]Aklillu E,Persson I,Bertilsson L,Johansson I,Rodrigues F,Ingelman-Sundberg M.Frequent distribution of ultrarapid metabolizers of debrisoquine in an ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles.J Pharmacol Exp Ther 1996;278:441-6.
[45]McLellan RA,Oscarson M,Seidegaa˚rd J,Evans DAP,Ingelman-Sundberg M.Frequent occurrence of CYP2D6 gene duplication in Saudi Arabians.Pharmacogenetics 1997;7:187-91.
[46]Fuselli S,Dupanloup I,Frigato E,Cruciani F,Scozzari R,Moral P,et al.Molecular diversity at the CYP2D6 locus in the Mediterranean region.Eur J Hum Genet 2004;12:916-24.
[47]Halling J,Petersen MS,Damkier P,Nielsen F,Grandjean P,Weihe P,et al.Polymorphism of CYP2D6,CYP2C19,CYP2C9 and CYP2C8 in the Faroese population.Eur J Clin Pharmacol 2005;61:491-7.
[48]Niewinski P,Orzechowska-Juzwenko K,Hurkacz M,Rzemislawska Z,Jaz´winska-Tarnawska E,Milejski P,et al.CYP2D6 extensive,intermediate,and poor phenotypes and genotypes in a Polish population.Eur J Clin Pharmacol 2002;58:533-5.
[49]Sistonen J,Sajantila A,Lao O,Corander J,Barbujani G,Fuselli S.CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure.Pharmacogenet Genomics 2007;17:93-101.
[50]Wan YJ,Poland RE,Han G,Konishi T,Zheng YP,Berman N,et al.Analysis of the CYP2D6 gene polymorphism and enzyme activity in African-Americans in southern California.Pharmacogenetics 2001;11:489-99.
[51]Weinshilboum R.Richard weinshilboum:pharmacogenetics:the future is here!Mol Interv 2003;3:118-22.
[52]Filipski KK,Mechanic LE,Long R,Freedman AN.Pharmacogenomics in oncology care.Front Genet 2014;5:73.
[53]Jin Y,Desta Z,Stearns V,Ward B,Ho H,Lee KH,et al. CYP2D6 genotype,antidepressant use,and tamoxifen metabolism during adjuvant breast cancer treatment.J Natl Cancer Inst 2005;97:30-9.
[54]Goetz MP,Rae JM,Suman VJ,Safgren SL,Ames MM,Visscher DW,et al.Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes.J Clin Oncol 2005;23:9312-8.
[55]Volpe DA,Tobin GA,Mellon RD,Katki AG,Parker RJ,Colatsky T,et al.Uniform assessment and ranking of opioid mu receptor binding constants for selected opioid drugs.Regul Toxicol Pharmacol 2011;59:385-90.
[56]Gasche Y,Daali Y,Fathi M,Chiappe A,Cottini S,Dayer P,et al.Codeine intoxication associated with ultrarapid CYP2D6 metabolism.N Engl J Med 2004;351:2827-31.
[57]Koren G,Cairns J,Chitayat D,Gaedigk A,Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother.Lancet 2006;368:704.
[58]Dale´n P,Frengell C,Dahl ML,Sjo¨qvist F.Quick onset of severe abdominal pain after codeine in an ultrarapid metabolizer of debrisoquine.Ther Drug Monit 1997;19:543-4.
[59]Ciszkowski C,Madadi P,Phillips MS,Lauwers AE,Koren G. Codeine,ultrarapid-metabolism genotype,and postoperative death.N Engl J Med 2009;361:827-8.
[60]Raimundo S,Fischer J,Eichelbaum M,Griese EU,Schwab M,Zanger UM.Elucidation of the genetic basis of the common‘intermediate metabolizer'phenotype for drug oxidation by CYP2D6.Pharmacogenetics 2000;10:577-81.
[61]Garcia-Barcelo´M,Chow LY,Chiu HF,Wing YK,Lee DT,Lam KL,et al.Genetic analysis of the CYP2D6 locus in a Hong Kong Chinese population.Clin Chem 2000;46:18-23.
[62]Miners JO,Birkett DJ.Cytochrome P4502C9:an enzyme of major importance in human drug metabolism.Br J Clin Pharmacol 1998;45:525-38.
[63]Wang D,Sun X,Gong Y,Gawronski BE,Langaee TY,Shahin MH,et al.CYP2C9 promoter variable number tandem repeat polymorphism regulates mRNA expression in human livers. Drug Metab Dispos 2012;40:884-91.
[64]Yasmeen F,Ghafoor MB,Khalid AW,Latif W,Mohsin S,Khaliq S.Analysis of CYP2C9 polymorphisms(*2 and*3)in warfarin therapy patients in Pakistan.Association of CYP2C9 polymorphisms(*2 and*3)with warfarin dose,age,PT and INR.J Thromb Thrombolysis 2015;40:218-24.
[65]Wei L,Locuson CW,Tracy TS.Polymorphic variants of CYP2C9:mechanisms involved in reduced catalytic activity. Mol Pharmacol 2007;72:1280-8.
[66]Azarpira N,Namazi S,Hendijani F,Banan M,Darai M. Investigation of allele and genotype frequencies of CYP2C9,CYP2C19 and VKORC1 in Iran.Pharmacol Rep 2010;62:740-6.
[67]Lee CR,Goldstein JA,Pieper JA.Cytochrome P450 2C9 polymorphisms:a comprehensive review of the in-vitro and human data.Pharmacogenetics 2002;12:251-63.
[68]Aynacioglu S,Brockmo¨ller J,Bauer S,Sachse C,Gu¨zelbey P,O¨ngen Z,et al.Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br J Clin Pharmacol 1999;48:409-15.
[69]Takahashi H,Echizen H.Pharmacogenetics of warfarin eliminationanditsclinicalimplications.ClinPharmacokinet 2001;40:587-603.
[70]Tabrizi AR,McGrath SD,Blinder MA,Buchman TG,Zehnbauer BA,Freeman BD.Extreme warfarin sensitivity in siblings associated with multiple cytochrome P450 polymorphisms.Am J Hematol 2001;67:144-6.
[71]Takahashi H,Kashima T,Nomizo Y,Muramoto N,Shimizu T,Nasu K,et al.Metabolism of warfarin enantiomers in Japanese patients with heart disease having different CYP2C9 and CYP2C19 genotypes.Clin Pharmacol Ther 1998;63:519-28.
[72]Aithal GP,Day CP,Kesteven PJ,Daly AK.Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications.Lancet 1999;353:717-9.
[73]Furuya H,Fernandez-Salguero P,Gregory W,Taber H,Steward A,Gonzalez FJ,et al.Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoinganticoagulationtherapy.Pharmacogenetics 1995;5:389-92.
[74]Mannucci PM.Genetic control of anticoagulation.Lancet 1999;353:688-9.
[75]Yasumori T,Chen LS,Li QH,Ueda M,Tsuzuki T,Goldstein JA,et al.Human CYP2C-mediated stereoselective phenytoin hydroxylation in Japanese:difference in chiral preference of CYP2C9 and CYP2C19.Biochem Pharmacol 1999;57:1297-303.
[76]Giancarlo GM,Venkatakrishnan K,Granda BW,von Moltke LL,Greenblatt DJ.Relative contributions of CYP2C9 and 2C19 tophenytoin4-hydroxylationinvitro:inhibitionbysulfaphenazole,omeprazole,and ticlopidine.Eur J Clin Pharmacol 2001;57:31-6.
[77]Steward DJ,Haining RL,Henne KR,Davis G,Rushmore TH,Trager WF,et al.Genetic association between sensitivity to warfarinandexpressionofCYP2C9*3.Pharmacogenetics 1997;7:361-7.
[78]McCrea JB,Cribb A,Rushmore T,Osborne B,Gillen L,Lo MW,et al.Phenotypic and genotypic investigations of a healthy volunteer deficient in the conversion of losartan to its active metabolite E-3174.Clin Pharmacol Ther 1999;65:348-52.
[79]Kidd RS,Straughn AB,Meyer MC,Blaisdell J,Goldstein JA,Dalton JT.Pharmacokinetics of chlorpheniramine,phenytoin,glipizide and nifedipine in an individual homozygous for the CYP2C9*3 allele.Pharmacogenetics 1999;9:71-80.
[80]Sullivan-Klose TH,Ghanayem BI,Bell DA,Zhang ZY,Kaminsky LS,Shenfleld GM,et al.The role of the CFP2C9-Leu 359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996;6:341-9.
[81]Jung F,Richardson TH,Raucy JL,Johnson EF.Diazepam metabolism by cDNA-expressed human 2C P450s:identification of P4502C18 and P4502C19 as low K(M)diazepam N-demethylases.Drug Metab Dispos 1997;25:133-9.
[82]Kaneko A,Lum JK,Yaviong J,Takahashi N,Ishizaki T,Bertilsson L,et al.High and variable frequencies of CYP2C19 mutations:medical consequences of poor drug metabolism in VanuatuandotherPacificislands.Pharmacogenetics 1999;9:581-90.
[83]Xie HG,Kim RB,Wood AJ,Stein CM.Molecular basis of ethnic differences in drug disposition and response.Annu Rev Pharmacol Toxicol 2001;41:815-50.
[84]De Morais S,Wilkinson GR,Blaisdell J,Nakamura K,Meyer UA,Goldstein JA.The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans.J Biol Chem 1994;269:15419-22.
[85]Roh HK,Dahl ML,Johansson I,Ingelman-Sundberg M,Cha YN,Bertilsson L.Debrisoquine and S-mephenytoin hydroxylation phenotypes and genotypes in a Korean population.Pharmacogenetics 1996;6:441-7.
[86]Kubota T,Chiba K,Ishizaki T.Genotyping of S-mephenytoin 4′-hydroxylation in an extended Japanese population.Clin Pharmacol Ther 1996;60:661-6.
[87]Xie HG,Huang SL,Xu ZH,Xiao ZS,He N,Zhou HH. Evidence for the effect of gender on activity of(S)-mephenytoin 4'-hydroxylase(CYP2C19)in a Chinese population.Pharmacogenetics 1997;7:115-9.
[88]Furuta T,Ohashi K,Kosuge K,Zhao XJ,Takashima M,Kimura M,et al.CYP2C19 genotype status and effect of omeprazole on intragastric pH in humans.Clin Pharmacol Ther 1999;65:552-61.
[89]Furuta T,Shirai N,Sugimoto M,Ohashi K,Ishizaki T. Pharmacogenomics of proton pump inhibitors.Pharmacogenomics 2004;5:181-202.
[90]Xia XM,Wang H.Gastroesophageal reflux disease relief in patients treated with rabeprazole 20 mg versus omeprazole 20 mg:a meta-analysis.Gastroenterol Res Pract 2013;2013:327571.
[91]Schwab M,Schaeffeler E,Klotz U,Treiber G.CYP2C19 polymorphism is a major predictor of treatment failure in white patients by use of lansoprazole-based quadruple therapy for eradicationofHelicobacterpylori.ClinPharmacolTher 2004;76:201-9.
[92]Furuta T,Ohashi K,Kamata T,Takashima M,Kosuge K,Kawasaki T,et al.Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer.Ann Intern Med 1998;129:1027-30.
[93]Orr ST,Ripp SL,Ballard TE,Henderson JL,Scott DO,Obach RS,et al.Mechanism-based inactivation(MBI)of cytochrome P450 enzymes:structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks.J Med Chem 2012;55:4896-933.
[94]Danielson P.The cytochrome P450 superfamily:biochemistry,evolution and drug metabolism in humans.Curr Drug Metab 2002;3:561-97.
[95]Lamba JK,Lin YS,Thummel K,Daly A,Watkins PB,Strom S,et al.Common allelic variants of cytochrome P4503A4 and their prevalenceindifferentpopulations.Pharmacogenetics 2002;12:121-32.
[96]Wrighton SA,Ring BJ,Watkins PB,VandenBranden M. Identification of a polymorphically expressed member of the humancytochromeP-450IIIfamily.MolPharmacol 1989;36:97-105.
[97]Aoyama T,Yamano S,Waxman D,Lapenson D,Meyer U,Fischer V,et al.Cytochrome P-450 hPCN3,a novel cytochrome P-450 IIIA gene product that is differentially expressed in adult human liver.cDNA and deduced amino acid sequence and distinct specificities of cDNA-expressed hPCN1 and hPCN3 for the metabolism of steroid hormones and cyclosporine.J Biol Chem 1989;264:10388-95.
[98]Gorski JC,Hall SD,Jones DR,VandenBranden M.Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A(CYP3A)subfamily.Biochem Pharmacol 1994;47:1643-53.
[99]Roy JN,Lajoie J,Zijenah LS,Barama A,Poirier C,Ward BJ,et al.CYP3A5 genetic polymorphisms in different ethnic populations.Drug Metab Dispos 2005;33:884-7.
[100]Kuehl P,Zhang J,Lin Y,Lamba J,Assem M,Schuetz J,et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression.Nat Genet 2001;27:383-91.
[101]Pandey AV,Flu¨ck CE,Mullis PE.Altered heme catabolism by heme oxygenase-1 caused by mutations in human NADPH cytochrome P450 reductase.Biochem Biophys Res Commun 2010;400:374-8.
[102]Wang M,Roberts DL,Paschke R,Shea TM,Masters BSS,Kim JJ.Three-dimensional structure of NADPH-cytochrome P450 reductase:prototype for FMN-and FAD-containing enzymes. Proc Natl Acad Sci U S A 1997;94:8411-6.
[103]Su¨ndermann A,Oostenbrink C.Molecular dynamics simulations give insight into the conformational change,complex formation,and electron transfer pathway for cytochrome P450 reductase. Protein Sci 2013;22:1183-95.
[104]Sandee D,Morrissey K,Agrawal V,Tam HK,Kramer MA,Tracy TS,et al.Effects of genetic variants of human P450 oxidoreductase on catalysis by CYP2D6 in vitro.Pharmacogenet Genomics 2010;20:677-86.
[105]Hart SN,Wang S,Nakamoto K,Wesselman C,Li Y,Zhong XB.Genetic polymorphisms in cytochrome P450 oxidoreductase influence microsomal P450-catalyzed drug metabolism.Pharmacogenet Genomics 2008;18:11-24.
[106]Jin Y,Chen M,Penning TM,Miller WL.Electron transfer by human wild-type and A287P mutant P450 oxidoreductase assessed by transient kinetics:functional basis of P450 oxidoreductase deficiency.Biochemi J 2015;468:25-31.
[107]Huang N,Pandey AV,Agrawal V,Reardon W,Lapunzina PD,Mowat D,et al.Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis.Am J Hum Genet 2005;76:729-49.
[108]Vasiliou V,Vasiliou K,Nebert DW.Human ATP-binding cassette(ABC)transporterfamily.HumGenomics 2009;3:281-90.
[109]Fujita T,Brown C,Carlson EJ,Taylor T,de la Cruz M,Johns SJ,et al.Functional analysis of polymorphisms in the organic anion transporter,SLC22A6(OAT1).Pharmacogenet Genomics 2005;15:201-9.
[110]Gottesman MM,Fojo T,Bates SE.Multidrug resistance in cancer:role of ATP-dependent transporters.Nat Rev Cancer 2002;2:48-58.
[111]Kioka N,Tsubota J,Kakehi Y,Komano T,Gottesman MM,Pastan I,et al.P-glycoprotein gene(MDR1)cDNA from human adrenal:normal P-glycoprotein carries Gly 185 with an altered pattern of multidrug resistance.Biochem Biophys Res Commun 1989;162:224-31.
[112]Hoffmeyer S,Burk O,Von Richter O,Arnold HP,Brockmo¨ller J,Johne A,et al.Functional polymorphisms of the human multidrug-resistance gene:multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo.Proc Natl Acad Sci U S A 2000;97:3473-8.
[113]Siegmund W,Ludwig K,Giessmann T,Dazert P,Schroeder E,Sperker B,et al.The effects of the human MDR1 genotype on the expression of duodenal P-glycoprotein and disposition of the probe drug talinolol.Clin Pharmacol Ther 2002;72:572-83.
[114]Gerloff T,Schaefer M,Johne A,Oselin K,Meisel C,Cascorbi I,et al.MDR1 genotypes do not influence the absorption of a single oral dose of 1 mg digoxin in healthy white males.Bri J Clin Pharmacol 2002;54:610-6.
[115]Tang K,Ngoi SM,Gwee PC,Chua JM,Lee EJ,Chong SS,et al. Distinct haplotype profiles and strong linkage disequilibrium at the MDR1 multidrug transporter gene locus in three ethnic Asian populations.Pharmacogenetics 2002;12:437-50.
[116]Yamauchi A,Ieiri I,Kataoka Y,Tanabe M,Nishizaki T,Oishi R,et al.Neurotoxicity induced by tacrolimus after liver transplantation:relation togenetic polymorphisms ofthe ABCB1(MDR1)gene.Transplantation 2002;74:571-2.
[117]Hennessy M,Spiers J.A primer on the mechanics of P-glycoproteinthemultidrugtransporter.PharmacolRes 2007;55:1-15.
[118]Sakaeda T,Nakamura T,Horinouchi M,Kakumoto M,Ohmoto N,Sakai T,et al.MDR1 genotype-related pharmacokinetics of digoxin after single oral administration in healthy Japanese subjects.Pharm Res 2001;18:1400-4.
[119]Verstuyft C,Schwab M,Schaeffeler E,Kerb R,Brinkmann U,Jaillon P,et al.Digoxin pharmacokinetics and MDR1 genetic polymorphisms.Eur J Clin Pharmacol 2003;58:809-12.
[120]Fung KL,Gottesman MM.A synonymous polymorphism in a common MDR1(ABCB1)haplotype shapes protein function. Biochim Biophys Acta 2009;1794:860-71.
[121]Kimchi-Sarfaty C,Oh JM,Kim IW,Sauna ZE,Calcagno AM,Ambudkar SV,et al.A‘‘silent”polymorphism in the MDR1 gene changes substrate specificity.Science 2007;315:525-8.
[122]Li J,Bluth MH.Pharmacogenomics of drug metabolizing enzymes and transporters:implications for cancer therapy. Pharmgenomics Pers Med 2011;4:11-33.
[123]Le´tourneau IJ,Slot AJ,Deeley RG,Cole SP.Mutational analysis of a highly conserved proline residue in MRP1,MRP2,and MRP3 reveals a partially conserved function.Drug Metab Dispos 2007;35:1372-9.
[124]Conseil G,Cole SP.Two polymorphic variants of ABCC1 selectively alter drug resistance and inhibitor sensitivity of the multidrug and organic anion transporter multidrug resistance protein 1.Drug Metab Dispos 2013;41:2187-96.
[125]Droz´dzik M,Bialecka M,Mysliwiec K,Honczarenko K,Stankiewicz J,Sych Z.Polymorphism in the P-glycoprotein drug transporter MDR1 gene:a possible link between environmental and genetic factors in Parkinson's disease.Pharmacogenetics 2003;13:259-63.
[126]Rothnie A,Callaghan R,Deeley RG,Cole SP.Role of GSH in estrone sulfate binding and translocation by the multidrug resistanceprotein1(MRP1/ABCC1).JBiolChem 2006;281:13906-14.
[127]Wojnowski L,Kulle B,Schirmer M,Schlu¨ter G,Schmidt A,Rosenberger A,et al.NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity.Circulation 2005;112:3754-62.
[128]Conrad S,Kauffmann HM,Ito K,Deeley RG,Cole SP,Schrenk D.Identification of human multidrug resistance protein 1(MRP1)mutations and characterization of a G671V substitution.J Hum Genet 2001;46:656-63.
[129]Meier Y,Pauli-Magnus C,Zanger UM,Klein K,Schaeffeler E,Nussler AK,et al.Interindividual variability of canalicular ATP-binding-cassette(ABC)-transporter expression in human liver. Hepatology 2006;44:62-74.
[130]Izzedine H,Hulot JS,Villard E,Goyenvalle C,Dominguez S,Ghosn J,et al.Association between ABCC2 gene haplotypes and tenofovir-inducedproximaltubulopathy.JInfectDis 2006;194:1481-91.
[131]Vogelgesang S,Kunert-Keil C,Cascorbi I,Mosyagin I,Schro¨der E,Runge U,et al.Expression of multidrug transporters in dysembryoplastic neuroepithelial tumors causing intractable epilepsy.Clin Neuropathol 2003;23:223-31.
[132]Krishnamurthy P,Schuetz J.Role of ABCG2/BCRP in biology and medicine.Annu Rev Pharmacol Toxicol 2006;46:381-410.
[133]Pedersen JM.ATP-Binding-Cassette transporters in biliary efflux and drug-induced liver injury.PhD thesis,Uppsala University.2013.
[134]Gradhand U,Kim RB.Pharmacogenomics of MRP transporters(ABCC1-5)andBCRP(ABCG2).DrugMetabRev 2008;40:317-54.
[135]Iida A,Saito S,Sekine A,Mishima C,Kitamura Y,Kondo K,et al.Catalog of 605 single-nucleotide polymorphisms(snps)among 13 genes encoding human atp-binding cassette transporters:ABCA4,ABCA7,ABCA8,ABCD1,ABCD3,ABCD4,ABCE1,ABCF1,ABCG1,ABCG2,ABCG4,ABCG5,and ABCG8.J Hum Genet 2002;47:285-310.
[136]Imai Y,Nakane M,Kage K,Tsukahara S,Ishikawa E,Tsuruo T,et al.C421A polymorphism in the human breast cancer resistance protein gene is associated with low expressionofq141kproteinandlow-leveldrugresistance1 supported in part by grants from the Ministry of Education,Culture,Sports,Science and Technology,the Ministry of Health,Labour and Welfare,Japan,and the Virtual Research Institute of Aging of Nippon Boehringer Ingelheim.Mol Cancer Ther 2002;1:611-6.
[137]Li J,Cusatis G,Brahmer J,Sparreboom A,Robey RW,Bates SE,et al.Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients.Cancer Biol Ther 2007;6:432-8.
[138]Niemi M.Role of OATP transporters in the disposition of drugs. Pharmacogenomics 2007;8:787-802.
[139]Sparreboom A,Gelderblom H,Marsh S,Ahluwalia R,Obach R,Principe P,et al.Diflomotecan pharmacokinetics in relation to ABCG2421C>Agenotype.ClinPharmacolTher 2004;76:38-44.
[140]Mwinyi J,Johne A,Bauer S,Roots I,Gerloff T.Evidence for inverse effects of OATP-C(SLC21A6)*5 and*1b haplotypes on pravastatin kinetics.Clin Pharmacol Ther 2004;75:415-21.
[141]Tirona RG,Leake BF,Merino G,Kim RB.Polymorphisms in OATP-C identification of multiple allelic variants associated with altered transport activity among European-and African-Americans.J Biol Chem 2001;276:35669-75.
[142]Niemi M,Backman JT,Kajosaari LI,Leathart JB,Neuvonen M,Daly AK,et al.Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics.Clin Pharmacol Ther 2005;77:468-78.
[143]Nishizato Y,Ieiri I,Suzuki H,Kimura M,Kawabata K,Hirota T,et al.Polymorphisms of OATP-C(SLC21A6)and OAT3(SLC22A8)genes:consequences for pravastatin pharmacokinetics.Clin Pharmacol Ther 2003;73:554-65.
[144]Meyer zu Schwabedissen HE,Kim RB.Hepatic OATP1B transporters and nuclear receptors PXR and CAR:interplay,regulation of drug disposition genes,and single nucleotide polymorphisms.Mol Pharm 2009;6:1644-61.
[145]Pasanen MK,Neuvonen M,Neuvonen PJ,Niemi M.SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid.Pharmacogenet Genomics 2006;16:873-9.
[146]Xiang X,Jada SR,Li HH,Fan L,San Tham L,Wong CI,et al. Pharmacogenetics of SLCO1B1 gene and the impact of*1b and *15 haplotypes on irinotecan disposition in Asian cancer patients.Pharmacogene Genomics 2006;16:683-91.
[147]Han JY,Lim HS,Shin ES,Yoo YK,Park YH,Lee JE,et al. Influence of the organic anion-transporting polypeptide 1B1(OATP1B1)polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancer.Lung cancer 2008;59:69-75.
[148]SEARCH Collaborative Group,Link E,Parish S,Armitage J,Bowman L,Heath S,et al.SLCO1B1 variants and statininduced myopathy—a genomewide study.N Engl J Med 2008;359:789-99.
[149]Hayer-Zillgen M,Bru¨ss M,Bo¨nisch H.Expression and pharmacological profile of the human organic cation transporters hOCT1,hOCT2 and hOCT3.Br J Pharmacol 2002;136:829-36.
[150]Dresser MJ,Leabman MK,Giacomini KM.Transporters involved in the elimination of drugs in the kidney:organic anion transporters and organic cation transporters.J Pharm Sci 2001;90:397-421.
[151]Somogyi A,Bochner F.Dose and concentration dependent effect of ranitidine on procainamide disposition and renal clearance in man.Br J Clin Pharmacol 1984;18:175-81.
[152]Kimura N,Okuda M,Inui K.Metformin transport by renal basolateral organic cation transporter hOCT2.Pharm Res 2005;22:255-9.
[153]Wang Z-J,Yin OQ,Tomlinson B,Chow MS.OCT2 polymorphisms and in-vivo renal functional consequence:studies with metforminandcimetidine.PharmacogenetGenomics 2008;18:637-45.
[154]Song IS,Shin HJ,Shim EJ,Jung IS,Kim WY,Shon JH,et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin.Clin Pharmacol Ther 2008;84:559-62.
[155]Tzvetkov MV,Vormfelde SV,Balen D,Meineke I,Schmidt T,Sehrt D,et al.The effects of genetic polymorphisms in the organic cation transporters OCT1,OCT2,and OCT3 on the renalclearanceofmetformin.ClinPharmacolTher 2009;86:299-306.
[156]Shu Y,Sheardown SA,Brown C,Owen RP,Zhang S,Castro RA,et al.Effect of genetic variation in the organic cation transporter 1(OCT1)on metformin action.J Clin Invest 2007;117:1422-31.
[157]Magro L,Moretti U,Leone R.Epidemiology and characteristics of adverse drug reactions caused by drug-drug interactions. Expert Opin Drug Saf 2012;11:83-94.
[158]Vandel S,Bertschy G,Bonin B,Nezelof S,Francois T,Vandel B,et al.Tricyclic antidepressant plasma levels after fluoxetine addition.Neuropsychobiology 1992;25:202-7.
[159]Woosley RL,Chen Y,Freiman JP,Gillis RA.Mechanism of the cardiotoxic actions of terfenadine.JAMA 1993;269:1532-6.
[160]Verbeurgt P,Mamiya T,Oesterheld J.How common are drug and gene interactions?Prevalence in a sample of 1143 patients with CYP2C9,CYP2C19 and CYP2D6 genotyping.Pharmacogenomics 2014;15:655-65.
[161]Phillips KA,Veenstra DL,Oren E,Lee JK,Sadee W.Potential role of pharmacogenomics in reducing adverse drug reactions:a systematic review.JAMA 2001;286:2270-9.
[162]Preskorn SH,Shah R,Neff M,Golbeck AL,Choi J.The potential for clinically significant drug-drug interactions involving the CYP 2D6 system:effects with fluoxetine and paroxetine versus sertraline.J Psychiatr Pract 2007;13:5-12.
[163]Takeuchi F,McGinnis R,Bourgeois S,Barnes C,Eriksson N,Soranzo N,et al.A genome-wide association study confirms VKORC1,CYP2C9,and CYP4F2 as principal genetic determinants of warfarin dose.PLoS Genet 2009;5:e1000433.
[164]Daly AK,King BP.Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003;13:247-52.
[165]Hyland R,Jones BC,Smith DA.Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole.Drug Metab Dispos 2003;31:540-7.
[166]Mikus G,Scholz IM,Weiss J.Pharmacogenomics of the triazole antifungalagentvoriconazole.Pharmacogenomics 2011;12:861-72.
[167]Mikus G,Scho¨wel V,Drzewinska M,Rengelshausen J,Ding R,Riedel KD,et al.Potent cytochrome P450 2C19 genotyperelated interaction between voriconazole and the cytochrome P4503A4inhibitorritonavir.ClinPharmacolTher 2006;80:126-35.
[168]Zhu L,Bru¨ggemann RJ,Uy J,Colbers A,Hruska MW,Chung E,et al.CYP2C19 genotype-dependent pharmacokinetic drug interaction between voriconazole and ritonavir-boosted atazanavir in healthy subjects.J Clin Pharmacol 2016.http://dx.doi. org/10.1002/jcph.798.
[169]Frelinger 3rd AL,Lee RD,Mulford DJ,Wu J,Nudurupati S,Nigam A,et al.A randomized,2-period,crossover design study to assess the effects of dexlansoprazole,lansoprazole,esomeprazole,and omeprazole on the steady-state pharmacokinetics and pharmacodynamics of clopidogrel in healthy volunteers.J Am Coll Cardiol 2012;59:1304-11.
[170]Lau WC,Waskell LA,Watkins PB,Neer CJ,Horowitz K,Hopp AS,et al.Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation a new drug-drug interaction.Circulation 2003;107:32-7.
[171]Rocca B,Petrucci G.Personalized medicine,pharmacogenetics,and clopidogrel:unraveling variability of response.Mol Interv 2010;10:12-9.
[172]Ma TK,Lam YY,Tan VP,Yan BP.Variability in response to clopidogrel:how important are pharmacogenetics and drug interactions?Br J Clin Pharmacol 2011;72:697-706.
[173]Sibbing D,Koch W,Gebhard D,Schuster T,Braun S,Stegherr J,et al.Cytochrome 2C19*17 allelic variant,platelet aggregation,bleeding events,and stent thrombosis in clopidogrel-treated patientswithcoronarystentplacement.Circulation 2010;121:512-8.
[174]Tiroch KA,Sibbing D,Koch W,Roosen-Runge T,Mehilli J,Scho¨mig A,et al.Protective effect of the CYP2C19*17 polymorphism with increased activation of clopidogrel on cardiovascular events.Am Heart J 2010;160:506-12.
[175]Clarke TA,Waskell LA.The metabolism of clopidogrel is catalyzed by human cytochrome P450 3A and is inhibited by atorvastatin.Drug Metab Dispos 2003;31:53-9.
[176]Kubica A,Kozinski M,Grzesk G,Fabiszak T,Navarese EP,Goch A.Genetic determinants of platelet response to clopidogrel.J Thromb Thrombolysis 2011;32:459-66.
[177]Neuvonen PJ,Niemi M,Backman JT.Drug interactions with lipid-lowering drugs:mechanisms and clinical relevance.Clin Pharmacol Ther 2006;80:565-81.
[178]Verschraagen M,Koks CH,Schellens JH,Beijnen JH.P-glycoprotein system as a determinant of drug interactions:the case of digoxin-verapamil.Pharmacol Res 1999;40:301-6.
[179]Westphal K,Weinbrenner A,Giessmann T,Stuhr M,Franke G,Zschiesche M,et al.Oral bioavailability of digoxin is enhanced by talinolol:Evidence for involvement of intestinal P-glycoprotein.Clin Pharmacol Ther 2000;68:6-12.
11 December 2015;revised 17 February 2016;accepted 8 March 2016
Available online 8 October 2016
Handled by Yixue Li
.
E-mail:chenshuqing@zju.edu.cn(Chen SQ).
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Construction and Analysis of Functional Networks in the Gut Microbiome of Type 2 Diabetes Patients
- Towards Personalized Intervention for Alzheimer’s Disease
- Long Non-coding RNAs and Their Roles in Non-small-cell Lung Cancer
- Oxford Nanopore MinION Sequencing and Genome Assembly
- Precision Medicine:What Challenges Are We Facing?
- Cancer Precision Medicine in China