Relief-PSO混合算法在基因微阵列特征选择中的应用
2016-11-02杜洪波董文娟
杜洪波,董文娟
(沈阳工业大学 理学院,辽宁 沈阳 110870)
Relief-PSO混合算法在基因微阵列特征选择中的应用
杜洪波,董文娟
(沈阳工业大学 理学院,辽宁 沈阳 110870)
在处理高维小样本、高冗余、高噪声的基因微阵列数据时,无法采用传统特征选择方法进行分析。针对该问题提出了一种结合Relief和粒子群优化算法(Relief-PSO)的混合特征选择方法。首先采用Relief预选滤除部分特征,然后以SVM-PSO封装算法选择出最优特征子集,采用典型的小样本高维公共微阵列数据测试算法。结果表明,总体分类精度不低于85%,与SVMRFE,SVMDEA特征选择算法进行了比较,基于Relief和PSO的混合特征选择算法精度较高,能够有效应用于基因微阵列数据的分析。
特征选择;Relief;PSO;基因微阵列
随着人类基因组测序计划的阶段性进展陆续完成,生命科学研究逐步迈进后基因组时代,以微阵列实验为代表的高通量检测技术日益兴起[1]。
由于DNA微阵列实验的成本高、实验次数少,以致基因表达谱数据呈现小样本特性;同时,实验测试表达的基因数量惊人,导致了基因表达谱数据呈现高维特性。在这种数据的高维小样本问题中,样本特征维数远远高于样本个数,传统的机器学习算法难以担负,给基因分析带来了极大的挑战[2-4]。
维数约简是处理该问题的主要途径,其包括特征抽取(FeatureExtraction)和特征选择(FeatureSelection)2种方式,前者是通过组合变化构造新的低维特征空间,后者是采用特定的评估标准选择最优特征子集,从而达到降维的目的[5]。相比而言,特征选择具有不改变原始特征空间、计算复杂度低、更为精确、易于理解等特点,适用于大规模数据处理。
通过判断是否有分类器的参与,可以将特征选择分为3大类:过滤式(Filter)、缠绕式(Wrapper)、嵌入式,前两种方法最为常用,区别在于学习过程是否独立[6-9]。Filter方法时间效率高,但正是由于其分别考量单个特征的特点,导致特征间存在的相关性被忽视,可能产生的分类模型与真实结果有较大偏差;相应的,Wrapper方法复杂度高,在速度上较慢,但选择出的规模较小的优化特征子集有利于关键特征的辨识。
Filter类典型特征选择算法包括Focus算法和Relief算法,后者具有效率高、不限制数据类型等优点,应用最为广泛。Wrapper类特征选择包括分类器和搜索算法两个组成部分,分类器中SVM广泛应用于wrapper特征选择,具有小样本学习、抗噪声性能强、学习效率高、推广性好等优点;搜索算法中粒子群优化算法(PSO)可以同SVM进行封装,具有卓越的全局搜索优化能力[10-11]。综上所述,采用Relief-PSO混合算法对基因微阵列特征选择问题进行研究与计算,首先利用Relief作为预选滤除部分特征,然后采用PSO进行搜索,SVM作为评估函数选择最优特征子集。
1 特征选择算法
Relief算法根据特征评估近距离样本的区分能力特征,即认为特征好的样本距离接近,特征差异大的样本距离疏远。其计算公式如下:

(1)
其中,H(x)为与样本x同类的最近相邻样本点;M(x)为与样本x异类的最近相邻样本点。
PSO算法模拟鸟群觅食行为,是一种基于群体协作的随机搜索算法,初始化随机粒子,每次迭代过程中粒子根据飞行经验调整速度向最优位置飞行,粒子速度与位置更新公式如下:
vij(t+1)=w·vij(t)+c1rand()·(pij(t)-
xij(t))+c2rand()·(pgj(t)-xij(t))
(2)
xij(t+1)=xij(t)+vij(t+1)
(3)
其中,t为迭代数;w为惯性权重;rand()为随机数,取值0~1之间;Pi为局部最优值,代表粒子i在搜索空间中所经过的最佳位置;Pg为全局最优值,代表整个粒子群在搜索空间中所经过的最佳位置;c1和c2为加速系数。
SVM算法通过非线性变换把输入样本映射到高维空间,寻找低VC维的最优分类超平面,将原约束问题转化为凸二次规划对偶表达式如下:
(4)
对不等式约束的二次函数求极值,有全局最优的唯一解,满足:
(5)
在式(5)的所有解中,非零样本为支持向量,通过线性组合的方式得到最有分类平面的权系数向量,分类阀值b*由式(4)中的约束条件解得,由此可得最优分类函数为
(6)
其中,sgn()为符号函数。

K(xi,xj)=<φ(xi)·(xj)>
(7)
封装算法中核函数采用的是径向基函数RBF,如下式所示:
(8)
通过寻优的方式调整容错惩罚系数C和内核参数γ从而影响分类精度。
为克服Relief算法不能去除冗余特征的缺点,采用了串联式组合特征选择算法Relief-PSO,该方法为两阶段特征选择算法:第一阶段,采用Relief得到特征权值,滤掉权值小于阈值的特征得到特征子集;第二阶段,以分类器准确率为特征子集的评估标准,采用PSO算法逐步去除冗余特征,寻找最优特征子集,算法流程如图1所示。
首先利用Relief算法对提取的特征进行筛选,保留了与目标类相关性较大的特征,然后利用PSO_SVM封装算法对特征子集和SVM核参数进行同步优化。Relief算法和PSO_SVM封装算法分别通过Weka软件和Matlab语言平台实现,最终得到最优容错惩罚系数C和内核参数γ分别为200和0.03。
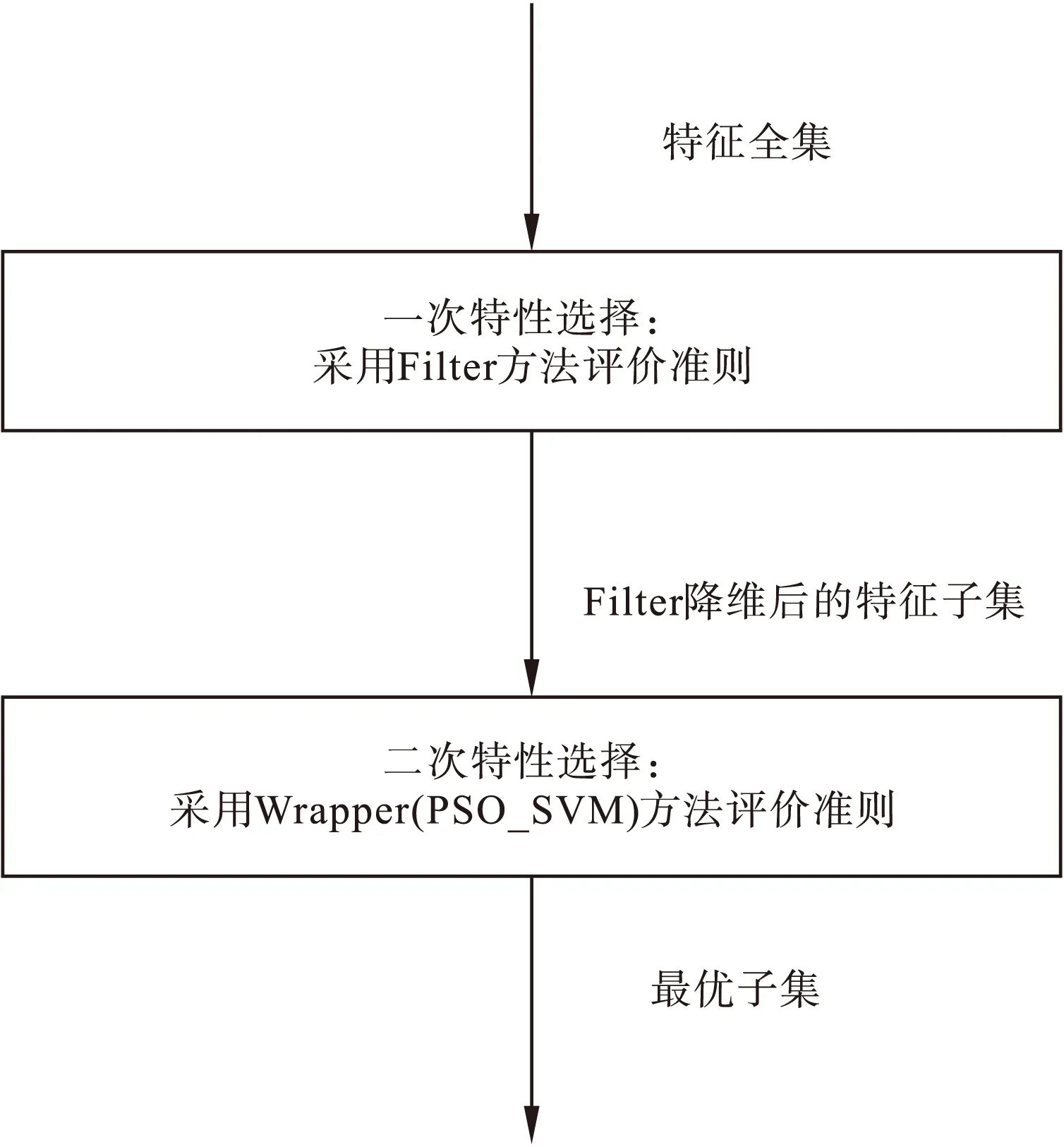
图1 算法流程
设PSO种群规模为N,既包含N个粒子,每个粒子的搜索空间为D,即粒子为D维向量,由样本维数决定,维数即数据集特征数,则基于PSO的基因选择算法描述如下:
Step1:Filter操作进行预选,剔除类无关噪声基因;
Step2:随机产生N个长度P的初始粒子群,粒子由候选基因子集组成,粒子群长度由Step1中预选基因子集决定;
Step3:计算当前粒子的使用度,支持SVM的交叉精度和选择的基因子集大小作为粒子优劣的参考标准;
Step4:更新局部个体最优和全局最优位置;
Step5:根据PSO算法更新每个粒子的位置;
Step6:产生新一代粒子群;
Step7:达到最大迭代数算法终止,否则跳到Step3。
2 实验数据
采用两个典型的高维小样本公共微阵列数据集来测试所提出特征选择方法,分别为:
1)结肠癌数据集(Colon)
该数据集搜集了结肠活组织样本中的表达值,数据集中包括62个结肠上皮细胞样本,基因表达水平通过使用约6 000个高密度寡核苷酸阵列来测量。经过测量表达水平的可信度选择,保留了2 000 个基因在40例腺癌(Cancer)和22例正常组织(Normal)的样本中的芯片表达数据集。
2)白血病数据集(Leukemia)
该数据集来自对两类急性白血病识别的芯片实验,基因表达水平为Aff.公司检测,包括47例急性淋巴增生性白血病(acutemyeloidleukemia,ALL)和25例急胜髓性白血病(acutemyeloidleukemia,AML)样本在7 129个基因中杂交结果。
实验所使用基因数据集由南洋理工大学的LiJ和LiuH收集,相关信息如表1所示。

表1 数据集相关信息
种群规模为50;最大迭代次数为200;结肠癌数据集w1=0.1,w2=0.2;白血病数据集w1=0.3,w2=0.4;粒子编码形式采用“0”、“1”制,其中“0”对应未被选择基因,“1”对应选择基因,解码过程中删除“0”对应基因,由“1”对应基因构成新的数据集,如结肠癌数据集有2 000个特征,则“0”、“1”编码长度为2 000。
3 结果与分析
首先利用Filter对基因表达谱数据的特征进行预选,使得数据特征的相关性得到了很大的提升。利用PSO_SVM分类器进行检验,可以完全对数据进行有效分类,在此过程中过滤法特征基因集包含分类所需要的基因,并没有去掉分类所用的信息基因,分类精度如表2所示。
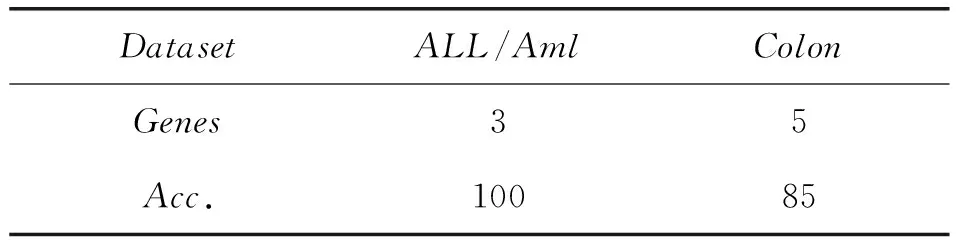
表2 实验结果
同时,为了进一步验证Relief-PSO特征选择方法的适用性,分别采用了以F-sore作为评价准则的Filter操作,与SVM-RFE、SVM-DEA方法进行了分类准确度对比,通过对比准确率来评价选择方法的优劣,对比结果如表3、图2所示。

表3 分类准确率对比结果
从表3中可以看出,在准确率(Acc.)方面,Relief-PSO在2个数据集上都要比SVM-RFE和SVM-DEA方法的分类准确率高,证明了所提出的混合特征选择算法能够解决基因微阵列特征选择问题,并取得更高的准确率。
4 结 论
针对高维小样本的基因微阵列特征选择问题,提出了一种混合Relief和PSO_SVM算法的混合特征选择方法,给出了算法流程,并应用在2个公共微阵列数据集上,对比结果表明,所提出的方法精度较高,能够满足基因微阵列特征选择的要求。

图2 分类准确率对比结果
[1]ZhangLJ,LiZJ.GeneSelectionforClassifyingMicroarrayDatausingGreyRelationAnalysis[C]//DiscoverScience2006.Barcelona,Spain:LNCS,2006,4265(1):378-382.
[2]ZhangLJ,LiZJ,ChenHW.AnEffectiveGeneSelectionMethodBasedonRelevanceAnalysisandDiscernibilityMatrix[C]//Pakdd2007.Nanjing,China:LNCS,4426:1088-1095.
[3]李瑶.基因芯片数据处理[M].北京:化学工业出版社,2006.
[4]CosminLazar,JonatanTaminau,StijinMeganck,etal.ASurveyonFilterTechniquesforFeatureSelectioninGeneExpressionMicroarrayAnalysis[J].IEEE,2012,9(4):11-19.
[5]InzaI,LarranagaP,BlancoR,etal.FilterversusgenewrapperapproachesinDNAmicroarraydomains[J].ArtificialIntelligenceinMedicine,2004,31(2):91-103.
[6]PodgorelecV,KokolP,StiglicB,etal.Decisiontrees:anoverviewandtheiruseinmedicine[J].JournalofMedicalSystems,2002,26(5):445-463.
[7]黄德双.基因表达谱数据挖掘方法研究[M].北京:科学出版社,2009.
[8]万洪强.应用于基因选择与癌症分类的微阵列数据分析[D].合肥:中国科技大学,2010.
[9]李颖新,软晓刚.基于支持向量机的癌症分类特征基因选取[J].计算机研究与发展,2005,42(10):324-330.
[10]ThomasJG,OlsonJM,TapscottSJ.AnEfficientandRebutStatisticalModelingApproachtoDiscoverDifferentiallyExpressedGenesUsingGenomicExpressionProfiles[J].GenomeResearch,2011,1(11):1227-1236.
[11]陆慧娟.基于基因表达数据的癌症分类算法研究[D].徐州:中国矿业大学,2012.
(责任编辑魏静敏校对张凯)
ApplicationofReliefandPSOHybridAlgorithmforGeneMicroarrayFeatureSelection
DUHong-bo,DONGWen-juan
(SchoolofScience,ShenyangUniversityofTechnology,Shenyang110870,LiaoningProvince)
Thetraditionalfeatureselectionmethodisunfitforthedataanalysisofgenemicroarraywithhighdimensionalsmallsample,highredundancyandhighnoise.Inthispaper,ahybridfeatureselectionalgorithmwasputforwardwhichiscombinedReliefwithparticleswarmoptimizationalgorithm(Relief-PSO).Firstly,afewcharacteristicswerepre-filteredwithRelief,andthen,theoptimalfeaturessubsetwaschosenbySVM-PSOencapsulationalgorithm.Finally,thetypicalhigh-dimensionalsmallsamplepublicmicroarraydatawasutilizedtotestthealgorithm.Theresultsshowthattheoverallclassificationaccuracyisnotlessthan85%.ThehybridfeatureselectionalgorithmhasahighprecisioncomparedwithSVMRFE,SVMDEAcharacteristicsselectionalgorithm,anditcanbeappliedtothegenemicroarraydataanalysismoreeffectively.
featureselection;Relief;PSO;genemicroarray
2016-05-11
杜洪波(1977-),男,吉林榆树人,副教授,硕士生导师,研究方向为数据挖掘、复杂网络。
董文娟(1990-),女,黑龙江黑河人,硕士研究生。
10.13888/j.cnki.jsie(ns).2016.03.016
TP391
A
1673-1603(2016)03-0267-05
