超高效液相色谱—串联质谱法分析鸡蛋中利巴韦林及其代谢物残留
2016-10-21齐凯汤晓艳王敏郑锌杨梦瑞周剑
齐凯 汤晓艳 王敏 郑锌 杨梦瑞 周剑

摘要 建立了超高效液相色谱串联质谱法同时快速测定鸡蛋中利巴韦林及其两种主要代谢物TCONH2和RTCOOH的分析检测方法。样品采用乙腈水(9∶1, V/V)提取,乙腈饱和正己烷除脂,C18結合GCB进行固相分散萃取除杂,Agilent ZORBAX SBAq色谱柱(100 mm × 3.0 mm,1.8 μm)分离,超高效液相色谱串联质谱测定。结果表明:利巴韦林、TCONH2和RTCOOH分别在2.0~200 μg/L, 0.5~200 μg/L, 5.0~200 μg/L浓度范围内,线性良好,相关系数R2>0.99,检出限分别为0.54, 0.09和1.54 μg/L,定量限分别为1.79, 0.31和5.13 μg/L。在5.0,10.0和50.0 μg/L加标水平下,利巴韦林和RTCOOH回收率分别为96.1%~99.6%和42.9%~58.3%;在0.5,2.0和5.0 μg/L加标水平下,TCONH2的回收率为75.9%~106.7%,相对标准偏差均为4.2%~12.7%。实际样品测定结果表明,本方法操作简单、快速、准确,能够满足鸡蛋中利巴韦林及其两种主要代谢物的分析检测。
关键词 超高效液相色谱串联质谱; 利巴韦林及其代谢物; 鸡蛋
1引言
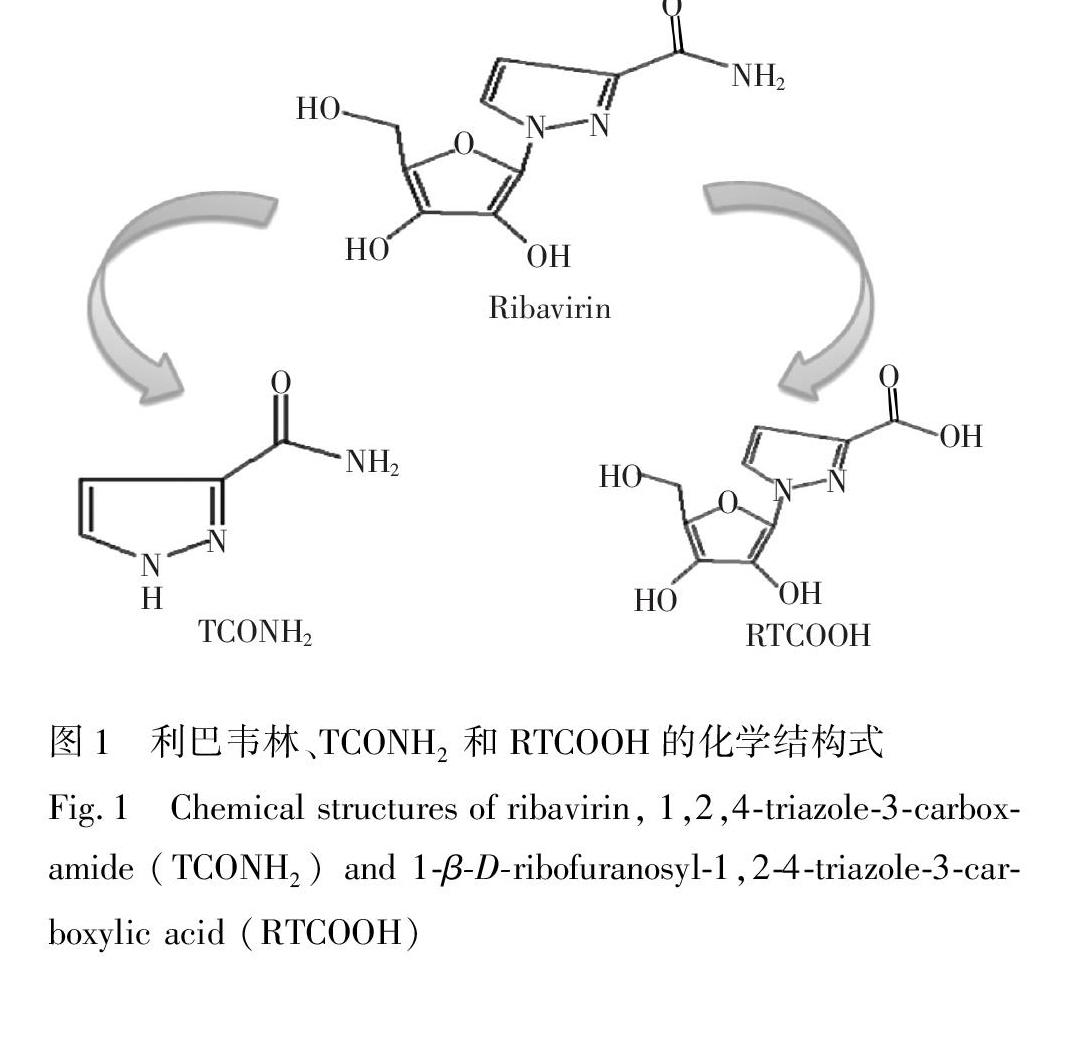
我国是禽蛋生产和消费大国,禽蛋产量多年来位居世界第一。禽类养殖过程中禽流感以及一些病毒性疾病一直是困扰养殖户的头等问题,为了减少损失,人用抗病毒药物利巴韦林曾被用于兽医临床用途。利巴韦林(1βDribofuranosyl1,2,4triazole3carboxamide)是一种人工合成的广谱抗病毒药物 [1 ],在动物细胞内被迅速代谢为TCONH2(1,2,4triazole3carboxamide)和 RTCOOH(1βDribofuranosyl1,2,4triazole3carboxylic acid)等相关代谢产物 [2 ],利巴韦林及其两种主要代谢物
图1利巴韦林、TCONH2和RTCOOH的化学结构式
Fig.1Chemical structures of ribavirin, 1,2,4triazole3carboxamide (TCONH2) and 1βDribofuranosyl1,24triazole3carboxylic acid (RTCOOH)化学结构式见图1。但利巴韦林具有一定的毒副作用,长期使用可使病毒产生不同程度的耐药性,诱导新型耐药病毒的出现,从而影响人类病毒疾病的防治,给人们的身体健康造成危害。因此,我国农业部于2005年发布第 [560 ]号公告,明确规定禁止在动物养殖过程中使用利巴韦林等人用抗病毒药物 [3 ]。产业调研发现,在我国禽类养殖过程中,违规使用利巴韦林等抗病毒性药物现象目前仍然十分严重,严重威胁禽类产品质量安全。因此有必要建立快速、准确、灵敏、高效的利巴韦林及其代谢物残留检测方法,为监管提供强有力的技术支撑。
目前,检测利巴韦林原型的方法有高效液相色谱法(HPLC) [4,5 ]、放射免疫法(RIA) [6 ]、毛细管电泳法(HPCE) [7 ]、液相色谱串联质谱法(HPLC/MS) [8~12 ]等,目标分析物主要涉及人血浆和动物源性食品。对于利巴韦林及其代谢物的检测,仅在动物血浆上有报道 [2 ],鸡蛋中利巴韦林及其代谢物的残留检测尚无相关报道。本研究采用超高效液相色谱串联质谱法(UPLCMS/MS)测定鸡蛋中利巴韦林及其两种主要代谢物TCONH2和RTCOOH的残留量,比较了不同前处理方法,优化了超高效液相色谱和质谱条件,同时进行了方法学评估,并应用于阳性鸡蛋样品中3种残留物检测分析。本方法操作简单、快速、准确,能够满足鸡蛋中利巴韦林及其主要代谢物的残留检测。
2实验部分
2.1仪器与试剂
ACQUITY UPLC超高效液相色谱(美国Waters公司);QTRAP 6500型三重四极杆线性离子阱复合质谱系统(美国AB SCIEX公司);TTLDCII型氮气浓缩仪(北京同泰联科技发展有限公司);MilliQ超纯水器(美国Millipore公司)。
利巴韦林标准品(德国Dr.Ehrenstorfer公司),纯度99.9%;13C5利巴韦林标准品(加拿大TRC公司),纯度99.9%;甲醇(HPLC级,德国MERCK公司);甲酸、正己烷(HPLC级,美国MREDA公司);乙腈(HPLC级,美国J.T.Baker公司);实验用水为MilliQ超纯水。
生鲜鸡蛋为本实验所用鸡蛋均为实验饲养蛋鸡所产,其中给药组为对蛋鸡连续3天饲喂30 mg/kg利巴韦林原药,停药1, 2, 5和7天后采集的鸡蛋样品。
2.2标准溶液配制
分别用甲醇配制浓度为100 mg/L的利巴韦林、TCONH2和RTCOOH标准储备液,
Symbolm@@ 20℃避光保存。移取0.1 mL利巴韦林、TCONH2和RTCOOH标准储备液混合,用空白鸡蛋提取净化液稀释成1.0 mg/L混合基质标准溶液,然后逐级稀释配制成0.5,1.0,2.0,5.0,10,20,50,100和200 μg/L共9种不同浓度梯度基质标准溶液。
2.3样品处理
准确称取鸡蛋液样品1.0 g(精确至0.01g)置于50 mL具塞离心管中,加入200 μg/L13C5利巴韦林标准溶液100 μL作为内标,加入10 mL乙腈水(9∶1, V/V)提取,5000 r/min离心10 min,转移上清液至50 mL离心管中。
在上清液中加入5 mL乙腈饱和的正己烷进行除脂,5000 r/min离心5 min,取下层溶液5 mL于10 mL离心管中,加入固相萃取填充料石墨化碳黑(GCB)和C18各50 mg进行净化,5000 r/min离心5 min, 取上清液氮气吹干,加1 mL纯水复溶,过0.22 μm滤膜后, 采用UPLCMS/MS测定。
2.4色谱条件
色谱柱: Agilent ZORBAX SBAq柱(100 mm×3.0 mm, 1.8 μm);流动相: A为0.1%(V/V)甲酸溶液,B相为甲醇;柱平衡時间1 min;梯度洗脱程序: 0~2.5 min,99% A;2.5~4.0 min,85% A;4.0~5.0 min,10% A;5.1~8.0 min,99% A;流速0.3 mL/min; 进样体积5 μL; 内标法定量。
2.5质谱条件
离子源: 电喷雾离子源(ESI);检测方式: 多重反应监测(MRM);扫描方式: 正离子扫描;碰撞气(CAD)压力为13.8 kPa;气帘气(CMR)压力为172.4 kPa;雾化气(GS1)压力为206.8 kPa;干燥气(GS2)压力为1.9 MPa;电喷雾电压(IS)为5500 V;离子化温度(TEM)为550℃;MRM定量离子对、去簇电压(DP)及碰撞电压(CE)见表1。
3结果与讨论
3.1提取溶剂的选择
利巴韦林属于强极性化合物,化学结构中包含酰胺基、羟基等基团,其油水分配系数lgP为
Symbolm@@ 2.06 [14 ],易溶于水和乙腈、甲醇等有机溶剂。目前, 针对动物组织中利巴韦林的提取剂主要有0.1%
甲酸乙腈溶液(1∶9, V/V)、0.1%甲酸20 mmol/L乙酸铵/甲醇溶液(1∶9, V/V)和乙腈水(9∶1, V/V)等 [15 ]。本实验比较了上述3种提取剂的提取效果。结果表明,0.1%甲酸乙腈溶液(1∶9, V/V)和0.1%甲酸20 mmol/L乙酸铵/甲醇溶液(1∶9, V/V)对利巴韦林及其代谢物并未达到理想的提取效果,且基质响应明显增大,而乙腈水(9∶1, V/V)对目标物具有很好的提取效果,且基质效应较前两者明显变小。分析原因,可能是酸性的提取液会对鸡蛋基质中具有与利巴韦林及其代谢物相似基团和结构的物质进行提取,从而导致质谱中基质响应变大,影响目标物分析,故本实验采用乙腈水溶液(9∶1, V/V)作为提取剂。
3.2净化条件的选择
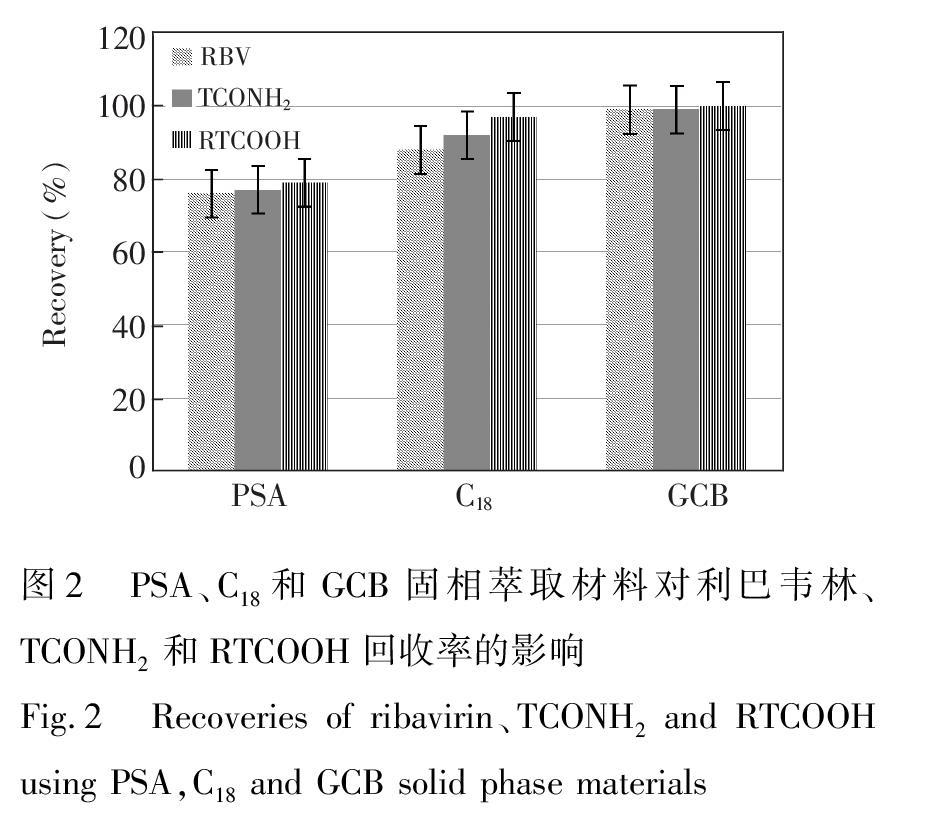
目前,对于生物组织及体液中利巴韦林的净化方式主要用固相萃取技术(SPE),采用磺酸基强阳离子(MCX)萃取柱、氨基(NH2)萃取柱、苯硼酸基弱阳离子(PBA)萃取柱和亲水亲脂平衡(HLB)萃取柱等 [16 ]。本方法应用QuEChERS快速样品前处理方法原理,采用乙腈饱和的正己烷去除溶于提取液中的油脂,用固相分散萃取填料除去脂溶性的杂质和色素等。实验中考察了3种固相萃取柱填料N丙基乙二胺(PSA)、C18和GCB对利巴韦林及其主要代谢物回收率的影响,如图2所示。结果表明,PSA对利巴韦林有较大的吸附作用,回收率在76%~79%之间,而C18和GCB对利巴韦林的吸附作用较小,回收率分别在88%~97%和99%~100%。分析原因发现,PSA在吸附基质中的色素、有机酸等生物分子时,对利巴韦林也产生了吸附作用,导致回收率偏低。因此,本研究选用C18和GCB用于固相萃取净化。此外,本方法与文献[16]报道的利巴韦林传统固相萃取法进行了比较,结果表明,采用本方法净化时,利巴韦林及其代谢物的回收率均高于固相萃取法,且重复性好,净化时间缩短,成本降低。
3.3色谱质谱条件的选择及优化
利巴韦林属于强极性化合物,在C18柱上保留性差。本实验采用能分离极性化合物的Agilent ZORBAX SBAq色谱柱(100 mm×3.0 mm, 1.8 μm)进行分离,发现利巴韦林在3.06 min处出峰,与RTCOOH达到了有效分离,与代谢物TCONH2分离度不够,但利巴韦林和TCONH2在后续质谱中电离生成的特征碎片离子对不同(m/z 245/113和m/z 113/96),根据不同特征离子对的质荷比即可准确定量利巴韦林原型和代谢物TCONH2。利巴韦林是核苷类似物,由于鸡蛋基质中含有大量的与利巴韦林及其代谢物分子量相同的核苷类似物,导致在提取的过程中,与目标物一同提取出来。这种基质在质谱中响应影响大,用目前的萃取方法难以除去,所以本实验采用液相色谱将基质与目标物进行了很好的分离,从而消除基质的影响。实验中发现,氮气吹干后的样品用甲醇溶液复溶,在同样的仪器条件下,利巴韦林及其代谢物在色谱柱中无保留,均在2 min前出峰。且利巴韦林及其代谢物和基质峰重合,由于基质效应影响大,很难对结果进行有效分析。所以本实验中均采用超纯水对处理后的样品复溶,基质在4.2 min处出峰,与3种目标物达到了有效的分离,保障了目标物准确的定性和定量分析。
本方法也同时考察了以0.1%甲酸甲醇作为流动相时色谱柱对目标物的分离效果。结果表明,在流动相中甲醇的比例超过30%时,利巴韦林及其代谢物在色谱柱上的保留性差,有双峰出现;随着甲醇比例减少,水相比例增加,利巴韦林峰形变好且与代谢物达到了有效分离;将水相比例维持在99%进行洗脱,此时得到的利巴韦林峰形较好,出峰时间有效避开了基质峰,排除了基质影响,结果见图3。
3.4线性范围、方法检出限和定量限
为了消除基质效应对测定结果的影响,本方法采用基质标准曲线对利巴韦林及其代谢物进行分析,以定量离子对和内标13C5利巴韦林的峰面积比为纵坐标(Y),以基质标准工作液浓度(0.5,1.0,2.0,5.0,10,20,50,100和200 μg/L)为横坐标(X)进行线性回归计算。结果表明,利巴韦林在2.0~200 μg/L浓度范围内线性关系良好,线性方程为Y=0.0669X+0.0608(相关系数R2=0.9997);TCONH2在0.5~200.0 μg/L浓度范围内线性关系良好,线性方程为Y=0.739X+3.79(相关系数R2=0.9931);RTCOOH在5.0~200.0 μg/L浓度范围内线性关系良好,线性方程为Y=0.0181X+0.0192(相关系数R2=0.9981)。以信噪比(S/N)确定方法的检出限(LOD)和定量限(LOQ),得到利巴韦林的LOD(S/N=3)为0.54 μg/L,LOQ(S/N=10)为1.79 μg/L;TCONH2的LOD为0.09 μg/L,LOQ为0.31 μg/L;RTCOOH的LOD为1.54 μg/L,LOQ为5.13 μg/L。
3.5方法的回收率和精密度
本实验采用在空白的鸡蛋中添加标准溶液的方法进行添加回收实验测定,内标法校正,每个浓度点平行测定6次,其加标回收率及精密度结果见表2。
3.6實际样品分析
应用本方法测定给药试验蛋鸡所产鸡蛋中利巴韦林原药及其代谢物。经检测分析,在停药5天内,鸡蛋样品中均检测出3种物质利巴韦林、TCONH2和RTCOOH,其中TCONH2残留量远大于利巴韦林原型和RTCOOH。随着停药时间延长,停药7天后,鸡蛋中均未检测到利巴韦林原药和代谢物RTCOOH,但TCONH2仍有残留检出,结果见表3。根据鸡蛋中利巴韦林及其代谢物残留消除的规律,确定较长时间段内残留在鸡蛋组织中的目标物为TCONH2,可作为蛋鸡养殖中监察违规使用利巴韦林的重要依据。
4结 论
建立了超高效液相色谱串联质谱检测鸡蛋中利巴韦林及其主要代谢物残留的方法,针对利巴韦林在组织中残留量低,转化率高的特点,分析了利巴韦林及其代谢物TCONH2、RTCOOH在鸡蛋中的残留情况。本方法很好地分离了利巴韦林及其代谢物和基质中的核苷类似物,消除了基质效应影响。实验中样品进样分析时间短,操作简单,分离度好,回收率高,定量限和精密度基本满足要求,为实际禽类养殖中利巴韦林的监管提供了有效的方法支撑。
References
1BoschM E, Sanchez A J R, Rojas F S, Ojeda C B. J. Pharm. Biomed. Anal., 2007, 45(2): 185-193
2Endres C J, Moss A M, Ke B, Govindarajan R, Choi D S, Messing R O, Unadkat J D. J. Pharmacol. Exp. Ther., 2009, 329: 387-398
3Ministry of Agricultrue. No. 560 Bulletin of the Ministry of Agricultrue of the People′s Republic of China. [20150112 ]
农业部. 中华人民共和国农业部公告第560号, [20150112 ]
4D′Avolio A, de Nicolo A, Simiele M, Turini S, Agnesod D, Boglione L, Cusato J, Baietto L, Cariti G, Calcagno A, Sciandra M, Di Perri G, Bonora S. J. Pharm. Biomed. Anal., 2012, 66: 376-380
5D′Avolio A, Ibanez A, Sciandra M, Siccardi M, de Requena D G, Bonora S, Di Perri G. J. Chromatogr. B, 2006, 835: 127-130
6Austin R K, Trefts P E, Hintz M, Connor J D, Antimicrob M F K. Agents Chemother., 1983, 24(5): 696-701
7WANG YongZhong, ZHANG GuoDong, L Bu. Chin. Hosp. Pharm. J., 2001, 1: 12-13
汪永忠, 张国栋, 吕 布. 中国医院药学杂志, 2001, 1: 12-13
8Melendez M, Rosario O, Zayas B, Rodriguez J F. J. Pharm. Biomed. Anal., 2009, 49(5): 1233-1240
9Yeh L T, Nguyen M, Lin C C. J. Chromatogr. Sci., 2003, 41: 255-260
10WU Fei, DING Li. Chin. J. Med. Inf., 2011, 8: 3532-3533
吴 飞, 丁 黎. 医学信息(中旬刊), 2011, 8: 3532-3533
11WANG WeiXia. Shandong J. Anim Hus & Vete Med., 2013, 7: 8-9
王维霞. 山东畜牧兽医, 2013, 7: 8-9
12Yeh L T, Nguyen M, Dadgostari S, Bu W, Lin C C. J. Pharm. Biomed. Anal., 2007, 43(3): 1057-1064
13ZHU YongLin, SHAO DeJia, JIANG TianMei, LU GuiPing, WU Qiong. Chin.J.Veterinary Drug., 2008, 7: 22-25
朱永林, 邵德佳, 蒋天梅, 陆桂萍, 吴 琼. 中国兽药杂志, 2008, 7: 22-25
14ZHU WeiXia, YANG JiZhou, YUAN Ping, SUN ChuanLian, WANG CaiJuan, SUN WuYong, ZHANG ShuSheng. Chinese Journal of Chromatography, 2013, 10: 934-938
祝伟霞, 杨冀州, 袁 萍, 孙传莲, 王彩娟, 孙武勇, 张书胜. 色谱, 2013, 10: 934-938
15Li W K, Luo S Y, Li S Y, Athill L, Wu A, Ray T, Zhou W, Ke J, Smith H T, Tse F L S. J. Chromatogr. B, 2007, 846(12): 57-68
16Berendsen B J A, Wegh R S, Essers M L, Stolker A A M, Weigel S. Anal. Bioanal. Chem., 2012, 402(4): 1611-1623
