后处理中锕系元素和贵金属元素光化学调价方法
2016-10-20李富海丁作铭林铭章叶国安
李富海,丁作铭,林铭章,2,*,何 辉,叶国安
1.中国科学技术大学核科学技术学院,安徽合肥 230027;
2.中国科学院核能安全技术研究所,安徽合肥 230031;
3.中国原子能科学研究院放射化学研究所,北京 102413
后处理中锕系元素和贵金属元素光化学调价方法
李富海1,丁作铭1,林铭章1,2,*,何 辉3,叶国安3
1.中国科学技术大学核科学技术学院,安徽合肥 230027;
2.中国科学院核能安全技术研究所,安徽合肥 230031;
3.中国原子能科学研究院放射化学研究所,北京 102413
PUREX流程是目前世界上唯一实现工业化应用的后处理流程,该流程的关键技术之一在于调控Pu、Np、Tc等元素的价态。传统上PUREX流程采用化学法调节元素价态,但化学调价有时会遇到困难,比如在硝酸体系下用化学试剂将Np调节至单一价态十分困难。和化学法相比,光化学调价具有方法简单、二次废物体积少、方便远程控制、对材料耐辐照要求不高等优点。将光化学调价和溶剂萃取结合很有潜力实现金属元素的高效分离,只依靠光化学方法将金属离子还原至单质状态也可以实现金属元素的分离和回收。此外,紫外光的照射还能在室温下显著促进UO2在硝酸溶液中的溶解。
光化学;调价;分离;UO2溶解
核能作为我国能源规划中用来替代煤、石油和天然气等化石燃料的主要能源形式,具有清洁、高效和经济等优点,在实现碳减排目标、环境保护和可持续发展方面有不可替代的作用[1]。然而核能的高速发展必然产生大量的乏燃料,乏燃料中含有大量U、Pu和次锕系元素(MA:Np、Am、Cm)以及大量的裂变产物(FP),这些乏燃料能否得到安全妥善的处理关系着核电能否健康、持续的发展下去。为实现核能的可持续发展战略,我国采取英、法、日、俄等国坚持的闭式燃料循环,即对乏燃料进行后处理,回收铀、钚,并通过再循环加以充分利用,以提高铀、钚的利用率,减少放射性废物的毒性和体积[2]。
目前世界上大多数的后处理厂采用的是以磷酸三丁酯(TBP)为萃取剂的PUREX循环流程。PUREX流程自1954年首次在美国的Savannah River后处理厂工业化以来沿用至今,现已经成为唯一大规模工业化的后处理流程。该流程的基本原理是利用不同价态铀、钚和裂变产物在TBP中分配比的差异,将铀、钚从乏燃料中分离回收,加以循环利用,以提高铀、钚的利用率。因此Np、Pu等元素价态的调节至关重要,将直接决定铀、钚和裂变产物的分离效率。
传统上PUREX流程中采用化学法调节元素价态,如以氨基磺酸亚铁作为将Pu(Ⅳ)还原为Pu(Ⅲ)的还原剂[3]。然而化学调价有时会遇到困难,比如在硝酸体系下用化学试剂将Np调节至单一价态十分困难,这导致用TBP萃取分离铀钚时,Np分布于有机相和水相,分配比较低[4-6],这就需要寻求其他方法实现镎与铀、钚的分离。大量研究表明,激光或紫外光诱导的光化学方法可以有效的调节Np的价态[5-7]。化学调价的另一个缺点是调价的选择性比较差,和化学法相比,通过控制反应参数光化学方法很有希望实现调价的选择性[4]。同时,光化学法还有方法简单的优点,往往只需添加还原剂或氧化剂。光化学法还可以有效地减少二次废物的体积,因为光子代替化学试剂参与了反应,不会增加溶液中盐的含量[7-8]。另外,光源可以布置在远离反应液的地方,因而方便远程控制,对材料耐辐照要求不高[5]。
实际上,早在1958年就有文献[9]报道了在高氯酸体系下Np(Ⅵ)被光还原至Np(Ⅴ);1965年印度研究人员发现当以含噻吩甲酰三氟丙酮的苯溶液从酸性溶液萃取Pu(Ⅵ)时,Pu(Ⅵ)被还原成了Pu(Ⅳ),而在黑暗条件下没有观察到类似现象[10],这说明发生的是光反应。1969年研究人员发现,在紫外光照射下,硝酸体系中的Pu(Ⅲ)和Pu(Ⅴ)都被氧化成了Pu(Ⅵ)[11]。到了20世纪70年代,随着激光技术的发展,光化学调价在后处理中的应用引起了研究者极大的兴趣。研究的热点集中在铀、钚、镎这三种元素,有些关注的是核燃料的光化学行为[11-15],有些关注的是Np的调价[5,16-17],还有些致力于发展能应用于后处理的光化学方法和技术[18-20]。然而这些研究仅仅描述了不同价态的这些元素的光化学行为,并未涉及光反应的机理。
在过去的20多年间,研究人员将更多的精力投入到对反应机理的探究上,如对铀酰离子()光解反应的研究[21-24],对Np的研究[4-5,7,25],对Pu的研究[25-28]。这些研究积累了大量实验数据,并取得了重要的研究结果,然而在不同条件下发生的光反应不同,其机理也不同。可能发生的光反应有以下三种:
1)光子作用于溶液中的阴离子或溶剂分子使之激发或电离,溶液中会同时产生氧化性自由基和还原性自由基,如果溶液中存在适当的自由基清除剂,以清除氧化性自由基或还原性自由基,剩余的还原性自由基或氧化性自由基就能使金属元素价态降低或升高[29]。
2)溶液中的金属离子在紫外区域具有电荷转移(charge transfer,CT)吸收带,在光子的作用下产生激发,使电子从阴离子或溶剂分子转移到金属离子上,从而将金属元素还原至低价态[30-33]。
3)在溶液中添加诸如TiO2的光催化材料,利用光照射从材料释放出来的电子还原金属离子[34]。
将光化学调价和溶剂萃取结合显然很有希望实现金属元素的高效分离和回收,另外一种实现金属元素分离回收的方法是将金属元素还原至单质状态,然后通过离心或过滤将其回收。若更进一步,通过控制反应条件实现先后将几种金属元素各自还原成单质,就能只靠光化学方法实现金属元素的分离和回收。此外,有研究表明,紫外光的照射还能在室温下显著促进UO2在硝酸溶液中的溶解,并且在一定范围内,UO2的量越大,其溶解速率越快。Sasaki等[4]认为,被低于350nm的紫外光激发处于激发态的硝酸根离子(*NO-3)起到了关键的作用。
该文将从光化学调价、光还原和分离以及紫外光照射促进UO2溶解三个方面,介绍后处理中锕系元素和铂族元素金属的光化学调价研究进展,并简单阐述发生的光反应的机理。
1 光化学调价
1.1 铀元素的光化学调价
铀元素作为裂变堆的燃料,是人们关注的核心元素之一。后处理中铀的价态是U(Ⅵ),在硝酸溶液中以铀酰离子()的形式存在[22]。一方面,铀酰离子形成的很多化合物有很高的光敏性[35];另一方面,激发态的铀酰离子(*)在退激发时会发射波长在460~600nm的光子[23],这为研究有*参与的反应过程额外提供了一种方便有效的检测手段[36]。因此的光化学行为,一直是研究的热点。
1)草酸做猝灭剂
草酸铀酰体系曾经是通用的化学光度计,一般通过测量分解的草酸量,也可以通过测量体系释放出的CO量,来确定体系吸收的光子量[41]。Heidt等[42]发现,草酸铀酰发生光解时一般不伴随U(Ⅵ)的还原,只有草酸被分解,如反应(1)、(2)所示。当然也有可能伴随U(Ⅵ)的还原,只是概率很低,如反应(3)所示。反应(1)和(2)发生的相对概率比(p1:p2)与体系的p H值直接相关[42],在p H值为0~7时,相对概率比随p H值升高而降低,CO的产额也逐步降低。而CO2的产额与p H值无关,当p H值从2增加到6时,U(Ⅳ)的产额(φ(U(Ⅳ))缓慢升高,但始终很低,仅从0.002 3升高到0.006 3,φ(U(Ⅳ))还与U(Ⅳ)自身的浓度有关[42-43]。

在紫外光激发下,U(Ⅵ)还可以通过电子转移反应(charge transfer to metal,CTTM)从C2O2-4得到一个电子,从而被还原为U(Ⅴ),同时C2O2-4分解生成CO2和·CO-2,如反应(4)所示。
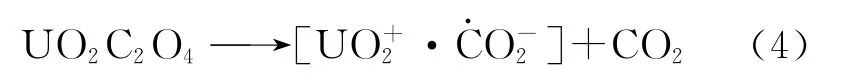
然而U(Ⅴ)并非经歧化反应生成U(Ⅳ)和U(Ⅵ),而是直接转化成U(Ⅵ)。在高p H条件下,U(Ⅴ)通过逆向电子转移反应失去一个电子被氧化为U(Ⅵ),同时有甲酸根离子生成(反应(5))。而在低p H条件下,HCO-2被CO取代(反应(6))。
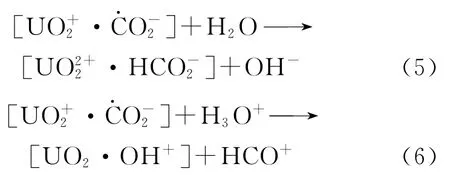
根据Heidt等[42]的研究结果,在草酸铀酰的体系下将U(Ⅵ)光还原至U(Ⅳ)效率极低。然而Mc Cleskey等[22]的研究结果却表明,在合适的条件下可以通过光解反应把U(Ⅵ)有效地还原成U(Ⅳ),并最终以UO2的形式沉淀下来。实验中以硝酸铀酰作为供体,为了消除NO-3的不利影响,所有实验加装滤光片截断λ<320nm的光。因为NO-3吸收300nm左右的紫外光分解产生强氧化性的氮氧化物(NOx),会抑制U(Ⅳ)的生成。p H=7,UO2的转化率达到96%。同样的条件,将p H调节为2.5,却几乎没有U(Ⅳ)生成,这说明溶液p H值的影响至关重要。Mc Cleskey等[22]也认为被光激发后通过电子转移反应生成了一种不稳定的U(Ⅴ)中间产物,然而他们认为这一中间产物可以通过第二次电子转移反应生成U(Ⅳ),如反应(7)所示。

当然,U(Ⅴ)也可以被·CO-2氧化重新生成U(Ⅵ),并同时生成CO和H2O或者HCOOH(反应(8)、(9))。

虽然很多文献[40,42,44]指出反应(9)会发生,但至今还没有从反应中分离出HCOOH[44],有研究者认为这是由于HCOOH很快被*分解的缘故[45]。但是在过量草酸盐存在的情况下,*的寿命小于20 ns,与HCOOH发生反应的几率大大降低,此时仍未能检测到HCOOH的生成不免令人困惑。但是CO在很多研究中都曾被观测到,因而Mc Cleskey认为,在U(Ⅴ)重新生成U(Ⅵ)的过程中起主导作用的是反应(8)。之所以造成前文所述Heidt和Mc Cleskey研究结果的差异,可能是由于实验条件并不相同,Mc Cleskey的所有实验结果都是在通氮气除氧后得到的,而Heidt并未提及这点,推测实验中并未除氧。
2)羧酸做猝灭剂
早期Heckler等[46]研究了和各种羧酸、二羧酸在p H为0.6~1.8及无氧条件下光解反应,认为在这一体系中发生的主要是脱羧反应,CO2是唯一的气体产物:
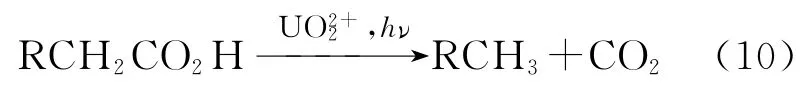
然而,对乙酸、丙酸、异丁酸等的电子自旋共振谱(ESR)研究结果[47]却表明,最初发生的光反应是夺氢反应,即*从与羧基直接相连的碳原子上夺走一个氢原子。Burrows等[36]认为,在浓溶液中起主导作用的是夺氢反应,而在稀溶液中是脱羧反应。Mc Cleskey等[22]发现,甲酸盐猝灭*时发生的是分子间的夺氢反应,而非分子内的电子转移反应。低p H(1.5、2.0、3.0)条件下,经λ>320nm光照射,U(Ⅵ)被还原为U(Ⅳ),紫外可见吸收谱确认了U(Ⅳ)的生成(图1)。
UV/Vis光谱分析没有得出铀酰离子-甲酸钠配合物存在的证据,同位素效应,以及反应速率在p H值低于和高于甲酸的p Ka(3.74)时无明显差别这些实验事实,都说明发生的反应是分子间反应。*从甲酸钠夺得一个氢原子后被还原,生成不稳定的U(Ⅴ)化合物,U(Ⅴ)化合物或者发生歧化反应,或者与高活性的自由基·COOH反应生成U(Ⅳ)和CO2,因而反应的最终产物只有U(Ⅳ)和CO2并没有CO,这与以前的研究结果一致[45]。此实验条件下,将反应后溶液的p H值调节到9,即可将U(Ⅳ)以UO2的形式沉淀下来,产额高达99.992%。并且保持浓度为0.1mol/L,把硝酸铀酰的质量从1g增大到150g并不会显著降低UO2的产额。Mc Cleskey等[22]的研究结果提供了一种新的、高效的将转化为UO2的方法。
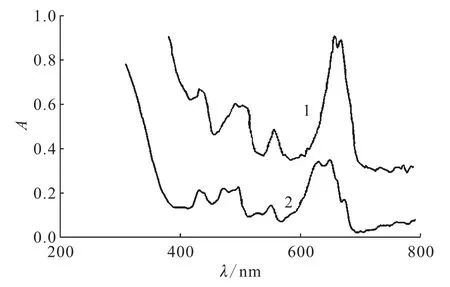
图1 铀酰-甲酸体系光解产物(1)与UCl4水溶液(2)的紫外可见吸收光谱Fig.1 UV/Vis spectra of U(Ⅳ)photoproduct(1)and UCl4dissolved in water(2)
3)醇做猝灭剂
U(Ⅵ)在各种醇存在的情况下可以很容易的被还原为U(Ⅳ),文献[48-51]测量了反应的Stern-Volmer常数Ksv,发现Ksv和通过测量φ(U(Ⅳ))得到的光反应的量子产额一致,并且乙醇等的绝对反应速率常数也和*在590nm处吸光度的衰减速率一致[52]。

(1)甲醇、乙醇、异丙醇和特丁醇的猝灭效果有如下关系式:甲醇<乙醇《异丙醇》特丁醇[50]。这与α-H的C—H键的键能呈负相关,即α-H越容易脱去,猝灭效果越好。而特丁醇没有α-H,因而猝灭效果远不如异丙醇。
(2)同位素效应。同位素效应的研究结果列入表1。由表1可知,当用D原子取代醇的α-H时,Ksv和φ(U(Ⅳ))都显著减小,这说明猝灭效果明显下降。
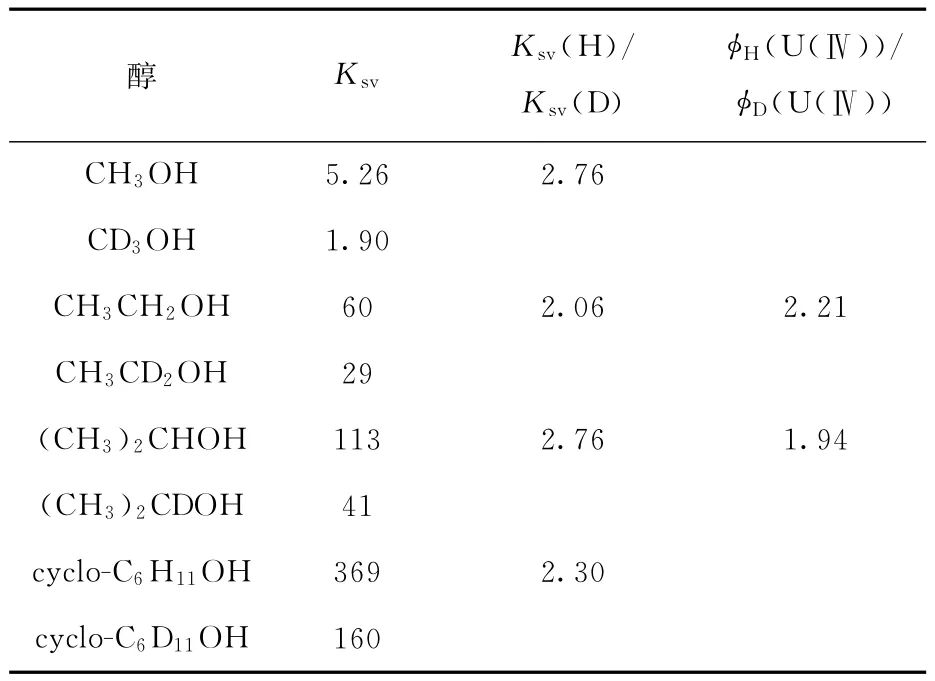
表1 同位素效应的研究结果Table 1 Results of research on isotope effects
(3)ESR研究表明,对伯醇(除了正丙醇),观测到了RC·HOH的ESR谱[47],这说明发生的是夺氢反应;对仲醇,也确认了C—C键裂解反应的存在,比如异丙醇有·CH3生成,特丁醇有·C2H5生成;而叔醇,以C—C键裂解反应为主;甲醇的结果比较有趣,Ledwith等[53]在液态甲醇自旋俘获方面的研究显示,被自旋俘获的主要自由基是CH3O·而非·CH2OH,而其它所有醇,被自旋俘获的主要自由基都是RC·HOH,这说明甲醇与*的作用是非典型的。
(4)乙醇的最终光解产物是乙醛,它是乙醇失掉α-H后生成的CH3C·HOH进一步被氧化的产物[48]。因而可以认为在铀酰-醇这一体系光解时,可能同时发生夺氢和C—C键裂解反应,这是一个竞争的过程,但对大多数醇而言,起主导作用的是夺氢反应。
Nagaishi等[23-24]发现*猝灭的反应速率常数kq和量子产额φ(U(Ⅳ))按照甲醇、乙醇和异丙醇的顺序递增[24],继续提高醇浓度到3mol/L,φ(U(Ⅳ))趋向于同一值。有氧条件下的φ(U(Ⅳ))大约只有无氧时的一半[23],说明反应是厌氧型。Nagaishi也认为在这一体系下发生的是夺氢反应;生成的UO+2不稳定,发生歧化反应生成U(Ⅳ)和U(Ⅵ)(反应(12));反应(11)生成的自由基RC·HOH具有还原性,可以直接将还原至U(Ⅴ)(反应(13))。

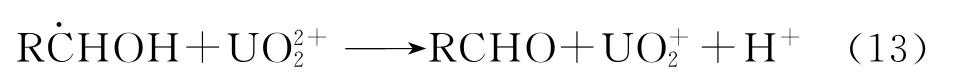
Salomone等[54]也研究了异丙醇做猝灭剂时硝酸铀酰中的光还原情况。p H调节为3,转化为UO2的转化率高达98%。至于反应机理,Salomone也认为发生的是夺氢反应。然而Salomone发现,NO-3的存在,不但不会抑制反而还会促进的光还原。这是由于NO-3和(CH3)2·COH反应生成了NO2-3,NO2-3能帮助还原(反应(14))。这与之前的文献报道大相径庭,因为一般认为NO-3吸收300nm的光发生光解,产生高氧化性的NOx,对的还原是不利的[55]。比如Mc Cleskey等[22]在研究硝酸铀酰光解时,为消除NO-3的不利影响,所有实验加装滤光片截断了λ<320nm的光。同样是硝酸铀酰体系,同样在酸性条件下,差别仅仅在猝灭剂不同,Salomone等[54]和Mc Cleskey等[22]却得到了截然不同的结果。NO-3到底在的光还原中起到什么样的作用,目前还不好定论,还需要更多研究来进行验证。
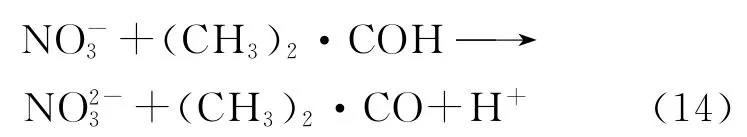
1.2 钚元素的光化学调价
在水溶液中,钚以Pu3+、Pu4+、PuO+2、PuO2+2四种氧化态离子存在,其中最稳定的是Pu4+。由于不同氧化态离子对之间的氧化还原电位值相差甚小,因此这四种氧化态离子能共存于水溶液中,但Pu O+2易歧化。
无任何添加剂时,可见光不影响硝酸溶液中Pu的价态,但在紫外光照射下,Pu(Ⅲ)和Pu(Ⅳ)都会被氧化到Pu(Ⅵ)[11]。Pu(Ⅲ)的氧化发生很快,并且反应速率随硝酸浓度(<3.5mol/L)增加而增大。段云富等[56]按照PUREX流程1BP的有关工艺条件,对Pu(Ⅲ)的光化学反应作了较为详细地研究。高压汞灯照射含Fe(Ⅱ)、肼和HNO3的Pu(Ⅲ)溶液,Pu(Ⅲ)能迅速而完全被氧化,60min后,溶液中的Pu(Ⅲ)即完全消失,Pu(Ⅳ)含量大于95%,Pu(Ⅵ)含量小于5%。光氧化分为三个阶段:(1)HNO3光解产物对Pu(Ⅲ)的氧化,但受到Fe(Ⅱ)、肼的强烈竞争,氧化速度缓慢;在这一阶段,Pu(Ⅲ)缓慢减少,Pu(Ⅳ)缓慢增加,没有Pu(Ⅵ)生成;(2)Pu(Ⅲ)的快速氧化过程,这是因为Fe(Ⅱ)和肼已基本耗尽,Pu(Ⅲ)迅速减少,Pu(Ⅳ)迅速增加,并开始产生少量Pu(Ⅵ);(3)产生的Pu(Ⅳ)经光歧化和光氧化产生少量Pu(Ⅵ)。
H2O2、乙醇和草酸等添加剂存在时,紫外光的照射会促进Pu(Ⅵ)向Pu(Ⅳ)的还原。H2O2还能使黑暗中硝酸溶液中的钚元素转化为Pu(Ⅲ),紫外光照射可以促进这一过程。
1.3 镎元素的光化学调价
在水溶液中,镎以Np(Ⅲ)、Np(Ⅳ)、Np(Ⅴ)和Np(Ⅵ)四种价态存在,但在氧化性酸性溶液中Np(Ⅲ)很容易被氧化到高价态,Np(Ⅶ)只存在于碱性溶液和固体状态下[8]。
研究表明[16,57],无添加剂时,紫外光照射使所有价态的Np都将转化为Np(Ⅴ),这一结果在HNO3浓度为0.1~6.0mol/L范围内都成立。然而,其它形式的光反应也可能发生,硝酸浓度和照射波长对反应都有影响,如表2[57]所示。这些反应在300nm光照射下也会进行,只是产额稍低。如果溶液中引入了氧气,会导致Np(Ⅵ)的积累。
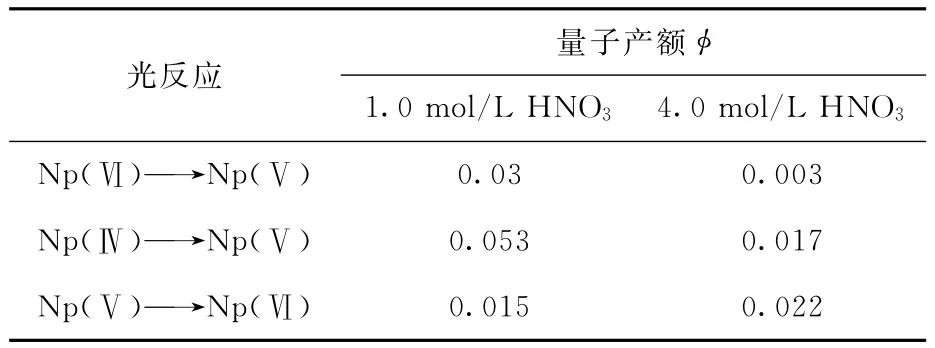
表2 硝酸溶液中Np在254nm光照射下几种光反应的量子产额[57]Table 2 Quantum yield of several photochemical reactions of Np in HNO3solution by 254nm light[57]
添加剂的引入,会显著改变Np的光化学行为。作为HNO2的清除剂,尿素可以使Np(Ⅴ)完全转化为Np(Ⅵ)[58],因为硝酸的光解产物HNO2有很强的还原性,能将Np(Ⅵ)还原到Np(Ⅴ)(反应(15))。Shilov等[59]的研究结果表明,1.8 mmol/L的Np(Ⅴ)在0.1mol/L尿素和3mol/L HNO3存在的条件下完全转化为Np(Ⅵ),反应进行完全需要5 h。H2O2能促进Np(Ⅳ)向Np(Ⅴ)的氧化,但硝酸浓度不应超过1.0mol/L。肼会促使Np(Ⅴ)转化为Np(Ⅳ),但反应不能进行完全。乙醇能促使Np(Ⅵ)还原至Np(Ⅴ),硝酸浓度为4.0mol/L时反应的量子产额为0.094[57]。

1.4 锝元素的光化学调价
99Tc是235U的主要裂变产物之一,其裂变产额高(6.13%),化学行为极复杂。一个现代化的核反应堆一年大约产生40kg99Tc[60],乏燃料后处理过程中大部分锝最终进入高放废液。在氧化和中性条件下,锝以Tc O-4的形式存在。由于99Tc的半衰期很长(2.13×105a),研究锝在地质环境中的迁移行为已经引起了人们极大的关注。TcO-4在环境中极易迁移,这是因为它带负电荷,很难被一般的矿物和岩石吸附。在美国的Hanford核废料储存地,大量易在土壤和地下水中迁移的99TcO-4已经成了河流最具风险的污染物。如果能把99TcO-4转换成为容易被岩石和矿物吸附的Tc(Ⅳ)O(OH)2,无疑将大大减轻99Tc在环境中的迁移[61]。

光催化剂(photocatalyst,PC)的引入也会改变Np的光化学行为。Fukasawa等[7]研究了Pt/ TiO2的引入对硝酸溶液中Np元素的光化学行为的影响。结果表明,只加入尿素时,90min后Np(Ⅴ)完全氧化到Np(Ⅵ);只加入PC时,PC不会产生任何影响,Np以同样速度完全转化为Np(Ⅴ);加入尿素时,PC能显著促进Np(Ⅵ)的生成,并最终将Np完全转化为Np(Ⅵ)。这是因为,反应中生成的HNO2会把Np(Ⅵ)还原成Np(Ⅴ),而尿素能清除HNO2,促进Np(Ⅵ)的积累;Np(Ⅴ)的氧化反应发生在PC的表面,光子从PC表面激发出一个自由电子e-,在PC上留下一个带正电的空穴h+,自由电子很快溶剂化形成水合电子,水合电子和NO-3反应掉,剩下的h+将Np(Ⅴ)氧化:
目前大量的相关研究集中在如何把Tc O-4还原为Tc(Ⅳ)(例如99TcO2·n H2O),然后把Tc(Ⅳ)吸附在固体表面或与玻璃、水泥、陶瓷这些材料混合,这样就可以把Tc从溶液中除去。然而,与以上这些材料混合的Tc(Ⅳ)最终都被重新氧化为了TcO-4。若能找到一种材料,可以通过化学成键结合并稳定还原低价Tc,则有望解决这一问题。基于这一思想,Burton-Pye等[62]引入了一种多金属氧酸盐(POM)α2-[P2W17O61]10-。在异丙醇存在的条件下,经紫外光激发,α2-[P2W17O61]10-与被还原的99Tc(Ⅴ)通过共价键结合,形成了一种稳定的99Tc(Ⅴ)O化合物99Tc(Ⅴ)O(α2-P2W17O61)7-。值得一提的是,在这一体系下太阳光也可以作为有效光源,只是反应速率慢得多。
2 还原分离
2.1 光化学方法与溶剂萃取结合分离金属元素
光化学调价具有传统化学调价不具有的选择性,然而单一体系下体现不出光化学调价的这一优点。在混合体系下,通过改变光源照射波长和时间、添加剂种类及浓度、阴离子种类及浓度、溶液p H值等反应参数,可以选择性地改变元素的价态。众所周知,溶剂萃取的效率与元素的价态息息相关,因而将光化学调价与溶剂萃取相结合,很有希望实现后处理中U、Pu、Np、Ln系元素等的分离。
1)U/Pu分离
最初PUREX流程中采用氨基磺酸亚铁作为还原剂还原Pu(Ⅳ),以实现U/Pu的萃取分离。但研究表明,硝酸铀(Ⅳ)作Pu(Ⅳ)的还原剂具有还原反萃完全、不向系统引入杂质、反应速率较快以及使不锈钢设备免受腐蚀等优点,现已慢慢取代氨基磺酸亚铁作为还原剂,广泛为各国轻水堆乏燃料后处理厂所采用。Toth等[14]研究了用光化学法还原U(Ⅵ)产生的U(Ⅳ)还原Pu(Ⅳ)的可行性,结果表明,该法可以实现铀钚的分离。整个过程可以看做是两步反应,在还原剂的帮助下,光解产生U(Ⅳ)(反应(18)),然后U(Ⅳ)将Pu(Ⅳ)还原(反应(19))。之所以不考虑Pu4+的光还原反应,是因为和光解反应相比,它的反应产额太低。

对比甲醇、乙醇、叔丁醇(Bu OH)、硝酸羟胺(HAN)和肼(N2H4)对光解的效果,结果表明BuOH最有效,N2H4的效果只有其十分之一,HAN则完全没效果。值得注意的是,只有N2H4存在时,U4+才是稳定的。为了既满足实验要求又能尽量减少引入添加剂的量,0.01mol/L Bu OH/0.01mol/L N2H4为最佳实验条件,此时U4+的生成速率为4×10-6mol/min。水相中模拟实验表明,c(HNO3)=1.0mol/L、c()= 1.0mol/L、c(Pu4+)=4×10-3mol/L时,所有的Pu4+都转化成了Pu3+,反应的量子产额为0.125~0.137。为了进一步验证此方法在PUREX流程中应用的可行性,又进行了两相(水相和有机相)模拟实验,结果表明,Pu4+很好地还原为Pu3+并进入水相,取得了很好的分离效果。
在Toth和Felker[14]的实验条件下,被光还原为U4+,而U4+又通过化学还原将Pu4+都还原为Pu3+,从结果来看,经过光反应后并没发生任何改变,而Pu4+都转化成了Pu3+,这也是光化学调价高选择性的体现。
2)Np/U分离
3)Np/Pu分离
Np/Pu分离也得到了研究人员足够的关注,Wada等[25]研究了硝酸溶液中Np/Pu混合体系的光化学行为。在进行光反应之前,用十倍量的HAN和N2H4将其预先调节为Pu(Ⅲ)和Np(Ⅴ)。研究了硝酸浓度和光功率密度对反应的影响,结果表明,Np(Ⅴ)保持不变,Pu(Ⅲ)会被氧化。光功率密度为0.5 W/cm2时,Pu(Ⅲ)迅速被氧化到Pu(Ⅳ),但Pu(Ⅳ)并不能稳定存在,开始照射后1min就快速被氧化为Pu(Ⅵ)。光功率密度为0.015 W/cm2时,Pu(Ⅲ)的氧化要慢得多,但随硝酸浓度增大而变快,当硝酸浓度为3.0mol/L时,照射10min即有95%的Pu(Ⅲ)氧化为Pu(Ⅳ),此后Pu(Ⅳ)保持稳定,此时非常适合用TBP萃取分离Np/Pu。关于光反应的机理,作者认为光激发产生的*NO-3起到了氧化Pu(Ⅲ)的作用。
Sasaki等[4]也进行了30%TBP/70%煤油对光化学调价后Np/Pu的萃取分离研究。和前面的研究结果一样,Pu(Ⅲ)被氧化到Pu(Ⅳ)和Pu(Ⅵ),但当硝酸浓度达到3.0mol/L时,Np(Ⅴ)会持续被还原到Np(Ⅳ),这与前面的结果[25]不同,或许是光功率密度不同的缘故。因而最适合进行Np/Pu萃取分离的实验条件为光功率密度0.15 W/cm2,硝酸浓度2.0mol/L。值得注意的是,对停止光照后的暗反应研究表明,Pu(Ⅵ)很快被还原到Pu(Ⅳ),部分Pu(Ⅳ)也被还原至Pu(Ⅲ),Np(Ⅴ)也很快被还原为Np(Ⅳ),因此萃取分离操作应在调价后立即进行或与调价同时进行。室温下的萃取实验表明,光照50min后87.1%的Pu进入了有机相,而99.8%的Np留在水相中,获得了不错的分离效果。
通过控制实验条件,光功率密度为0.15 W/ cm2,c(HNO3)=2.0mol/L,文献[4]选择性地将Pu(Ⅲ)氧化到Pu(Ⅳ),同时Np(Ⅴ)保持不变,结合溶剂萃取实现了Np/Pu的分离。这只是单级操作的结果,若进行多级操作,理应能获得更好的分离效率。
4)Ln系元素分离
三价镧系元素的结构与性质非常相似,实现它们的相互分离十分困难。然而它们的光学性质却有一定差别,这一点可用来实现它们的相互分离。最早将光化学方法应用于Ln系元素分离的文献报道出现在1977年,Donohue[65]选择性地将Eu3+光还原为Eu2+,结合沉淀法,成功把Eu从其它Ln系元素中分离出来。Eu是所有Ln系元素中最容易被还原的,当照射光波长正好处于Eu3+的CT吸收带时[29,66],Eu3+很容易还原到Eu2+(反应(20)),而EuSO4不溶,因而可以加入硫酸盐将Eu2+沉淀分离出来。另外一种容易光还原的Ln系元素是Sm[67]。

将光照产生的沉淀清洗后溶于浓硝酸,通过光谱分析不同元素含量,Eu/Sm/Ho照射前后以及沉淀溶于浓硝酸的光谱示于图2。最终超过90%的Eu3+被还原;Eu的分离因子β如下式:
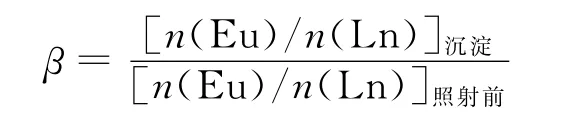
β对不同的Ln系元素有很大差别,从β(Eu/Pr)=1变化到β(Eu/Tm)=200,Donohue[65]认为离子半径越接近发生共沉淀的可能性越大。
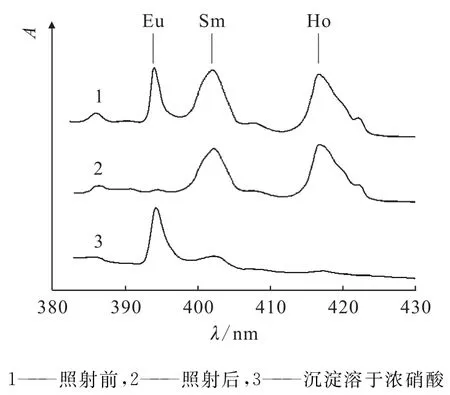
图2 Eu/Sm/Ho光谱分析Fig.2 Absorption spectra for equimolar Eu/Sm/Ho mixture
Tsushima等[68-69]探究了Ln(NO3)3-(NH4)2SO4-HNO3(Ln:Nd、Sm、Eu、Gd)体系下通过光还原和化学沉淀实现Ln系元素分离的可行性。结果发现:可以选择性的把Nd3+、Sm3+、Eu3+都还原到+2价,并通过硫酸盐的形式沉淀出来,而94.2%的Gd仍以Gd3+形式留在溶液中,这就实现了Gd与Nd、Sm、Eu的分离。把Gd换成La,实验结果很不一样,只有8.2%的La元素留在溶液中[68]。实验中采用的光源是超高压汞灯(350~410nm),Tsushima认为这一光源只能使Nd3+(354.0nm)、Sm3+(401.5nm)、Eu3+(394.2nm)还原,而不能使Gd3+(272.8nm)还原,并且Nd2+、Sm2+、Eu2+的离子半径比Gd3+大得多,因而共沉淀现象不明显[69],而La3+与Nd2+、Sm2+、Eu2+的离子半径很接近,发生了共沉淀现象[68]。故该工作还是利用Ln系元素CT吸收带的不同,通过控制光源照射波长,使其选择性地还原,从而达到分离的目的。
2.2 铂族元素金属的光还原
前面提到,光化学方法还可以通过将金属离子还原至单质,从而实现金属元素的分离。然而,U、Pu、Np、Am这些元素往往处于高氧化态,将其直接还原至单质十分困难。高放废液中除了这些元素之外,还有如Pd、Ru、Rh等铂族金属元素,它们比较容易还原至单质,这引起了一些研究人员的兴趣,并已有相关文献报道。有效地提取高放废液中Pd、Ru、Rh等贵重金属,既能够充分地利用资源,具有重要的经济价值,同时还可以减轻环境负担,符合循环经济的要求。另外,从高放废液的玻璃固化角度来看,由于铂族金属元素的熔点高于玻璃固化的温度,对于设备的运转会产生不利的影响[70]。因此,为了使玻璃固化工厂能够顺利运转,对于铂族元素的分离回收技术的开发尤为重要。
1)单一体系
Song等[71]比较了在Nd:YAG激光(355nm)照射下,不同自由基清除剂(草酸、甲酸、乙醇)在不同酸性溶液(硝酸、高氯酸、盐酸)中PdCl2光还原至钯单质的情况,并得到光还原PdCl2的最优方案。结果表明,以硝酸为溶剂,草酸为自由基清除剂可最大程度将PdCl2还原成钯单质。X射线衍射(XRD)分析表明,还原得到的沉淀物几乎为纯的钯单质。证实了使用其他方法需要很复杂步骤才能制得的高纯度钯单质,在使用光还原的方法时仅需一步就可制得,这极大地简化了制备钯单质的工艺流程。
Harada等[72]采用聚乙烯吡咯烷酮(PVP)为分散剂,成功用光还原法制备出平均粒径仅为1.3nm的钌。光照120min后Ru3+的特征吸收峰消失,表明Ru3+已全部还原成钌单质。Ohtaki等[73]采用乙醇或异丙醇为自由基清除剂,成功将RhCl3光还原至铑单质。该研究还解释了Rh3+在溶液中的光还原机理:
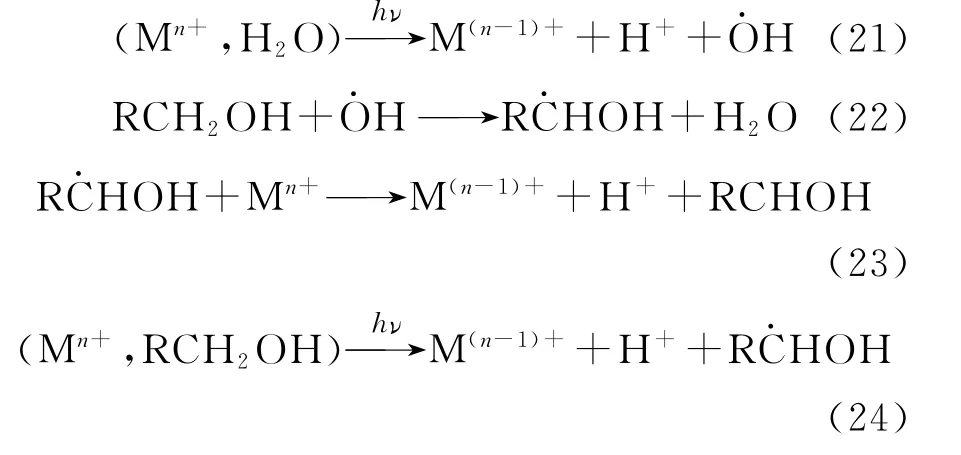
首先,水合铑离子受光激发被还原成低价铑离子,同时生成质子和自由基(反应(21));随后,溶液中的醇与反应生成RHOH,RHOH与铑离子进一步反应,最终将其还原成单质,如反应(22)、(23)所示。铑离子还可以直接与醇配位,经紫外光激发,直接发生夺氢反应(反应(24))。笔者认为在醇类自由基清除剂存在的条件下起主导作用的是反应(24),因为醇的α-H相比水中的氢原子更易被夺去。
综上可知,光化学方法可将铂族金属元素直接光还原至单质。故反应不需额外添加能使之沉淀的试剂,这就使工艺流程得到简化。
2)混合体系
混合体系下,金属离子的光化学行为变得复杂,要实现它们之间的相互还原分离十分困难,往往还要结合TiO2光催化技术。能带理论认为,当TiO2受到的外界光源的照射能量等于或超过TiO2的带隙能(3.0~3.2 e V)时,便会在导带和价带上分别产生大量的电子(e-)和空穴(h+)对,电子和空穴会同时向TiO2的表面迁移[74-75],在TiO2表面便与吸附在其表面的物质发生反应。所以,若能清除氧化性的空穴,便可利用光照从TiO2表面释放出来的电子还原金属离子。
Kriek等[76]利用光化学方法,通过调节p H值实现了PdCl2-RhCl3-H2Pt Cl6混合体系金属元素的分离:调节体系p H=11.9时,Pd2+和Rh3+被吸附在TiO2表面,而Pt4+仍留在溶液中,这将Pt4+与Pd2+和Rh3+分离;随后调节体系p H= 3.1,Pd2+被光还原,这使Pd2+与Rh3+分离。该研究还提出可尝试使用日光辐照铂族金属溶液,实现其还原分离,若能实现将大大节约能源。
Borgarello等[77]使用模拟日光辐照TiO2分散体与PdCl2和RhCl3的混合溶液,Pd2+和Rh3+被还原,并沉积在TiO2表面。结果表明,氧气会抑制Rh3+的还原,但对于Pd2+的还原没有影响,因而可以实现二者的分离。该研究还成功使用光还原法结合TiO2光催化材料,实现了AuCl3-H2PtCl6-RhCl3混合体系金属元素的分离:调节p H=0时,Au3+极易被光还原并沉积在TiO2表面,实现了Au与Pt、Rh的分离;调节p H=2.7时,只有Pt4+被还原,这可使Pt、Rh分离。
综上可知,光还原法在去除废液中的贵金属离子工艺中显示了独到的优势,通过控制反应参数,可以选择性地使一种或两种贵金属离子先后光还原,从而实现它们的分离。
3 紫外光照射促进UO2溶解
3.1 反应机理
UO2在硝酸中的溶解是后处理工艺中最关键的技术流程之一,研究如何提高UO2在硝酸中的溶解度具有十分重要的意义。早期研究结果表明,温度对UO2在硝酸中的溶解速率影响较大,UO2可以溶于热的浓硝酸中。Ikeda等[78-79]使用17O标记的UO2粉体,研究了UO2在硝酸体系下的溶解机理。他们通过对核磁共振数据的分析,证明了UO2的溶解是基于溶剂(如硝酸)和UO2粉体之间存在电子转移的氧化还原反应。反应方程式如下:
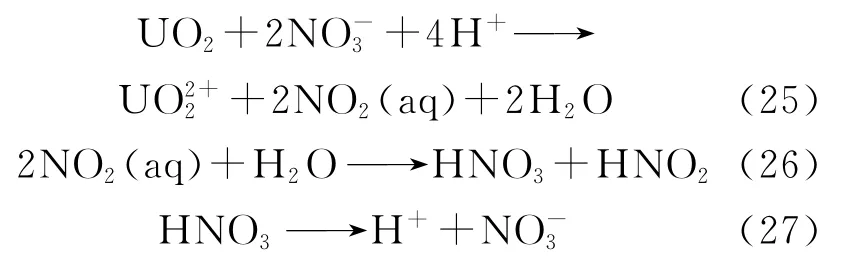
总反应方程式为:

随后,生成的亚硝酸也参与UO2溶解:
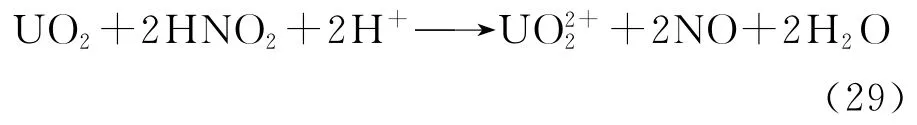
近年来,为了改良UO2溶解工艺,多数研究倾向于寻找在室温下使UO2快速溶解的方法。文献[4]发现,用汞灯照射含UO2粉体的硝酸溶液可促进UO2粉体溶解。他们认为在紫外光照射条件下,溶液中的NO-3会激发至激发态*NO-3,UO2与*NO-3发生电子转移反应,从而溶解于硝酸溶液中。反应(28)可改写成:
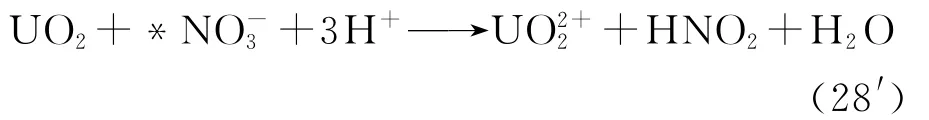
Kim等[80-81]发现,在黑暗环境下几乎不溶的UO2经紫外光照射仅7 h即溶于硝酸,这说明光照可明显增加烧结的UO2颗粒在硝酸中的溶解速率。并且在UO2溶解的同时,观测到了H2O2和NO-2的生成。与文献[4]不同的是,Kim认为反应式(28′)中的NO-2并非直接由*NO-3生成,而是先由*NO-3转化成NO2,再由NO2生成:

O-与溶液中的H+结合生成·OH自由基,后生成H2O2:

因此,紫外光照射条件下UO2在硝酸中溶解的反应方程式如下:

3.2 影响因素
由上述反应机理可知,有两种可以有效提高光化学法溶解UO2效率的方法:一是提高溶液中氧化性基团(如*NO-3、NO-2)的浓度,这可以通过增加光照量和提高硝酸浓度来实现;二是提高UO2的比表面积,使UO2能更容易接触到溶液中的氧化性基团,这可以通过使用氧化剂对UO2颗粒进行表面刻蚀实现。下列实验结果分别证实了以上观点。
1)光照量的影响
文献[4]研究了光照量分别为0、0.7、1.3 W/ cm2时UO2粉体溶解率系数(V,mol/(cm2·min))的变化规律,V的计算公式如下:

其中:ΔD0为溶解曲线的初始斜率,mol/min;S0为UO2粉体的比表面积,m2/g;m为UO2粉体的质量,mg。结果表明,V1.3与V0.7的比值可达3.3,而V1.3与V0的比值可达14.4,这意味着增强光照量可显著提高UO2的溶解速率。
2)硝酸浓度的影响
文献[4]发现,硝酸浓度为6mol/L和3mol/L时,它们的溶解率系数V的比值V6/V3为40.0,而V6与V1的比值高达3 590。从上述结果可看出,与增加光照量相比,增加硝酸浓度能更有效地提高UO2溶解的速率。
3)UO2质量的影响
文献[4]研究发现,当m(UO2)为1~100 mg时,m(UO2)=100 mg时溶解率系数最高。所以,适当增加UO2粉体的质量也可有效地提高UO2溶解的速率。
4)其它因素的影响
Kim等[81]向硝酸反应液中添加了Cs、Sr、Zr、Ru、Mo和Nd等金属离子来模拟放射性废液。结果表明,UO2颗粒在模拟废液中的溶解速率远大于在硝酸溶液中的溶解速率,且模拟废液中只有Ru离子和Mo离子可影响UO2颗粒的溶解速率。研究还发现,Mo(Ⅵ)只有在紫外光照射的条件下才可促进UO2颗粒的溶解。Kim推测,这里起作用的是激发态的*Mo(Ⅵ)。此外,粉末状的UO2在硝酸中的溶解速率是UO2颗粒的3倍。这是因为,与UO2粉体相比,UO2颗粒由于比表面积很小,故很难溶解于硝酸中。因此,为使UO2易溶于硝酸,应适当地增大UO2的比表面积。
上述研究结果表明,紫外光的照射可有效促进UO2在硝酸中的溶解。在这一过程中起关键作用的是经紫外光激发处于激发态的*NO-3,它和UO2之间存在电子转移的氧化还原反应,使UO2溶解。
4 结论与展望
综上所述,和化学调价相比,光化学调价因具有方法简单、二次废物体积少、方便远程控制、对材料耐辐照要求不高、高选择性等优点,很有希望应用于后处理中Pu、Np等元素的调价。大量的研究表明,光化学法可以有效地调节U、Pu、Np、Tc等元素的价态,光化学调价和溶剂萃取相结合可以实现U/Pu、U/Np、Np/Pu和Ln系元素的高效分离。只依靠光化学方法,通过控制p H值等反应参数也可以实现混合体系中铂族等贵金属元素的分离回收。此外,紫外光照射可以在室温下显著促进UO2在硝酸溶液中的溶解。
从实际应用角度来看,前人的研究都是在实验室中完成的模拟实验,模拟条件过于理想,未结合实际工艺条件。很重要的一点,在真正的乏燃料后处理流程中,很多元素都是有放射性的,水溶液一直在辐解,辐解产物中既有水合电子等强还原性的产物,也有·OH等强氧化性的产物,这些辐解产物一定会影响金属元素的价态。另外,真正的后处理溶液中都是多种元素同时存在,此时要想实现光化学的选择性调价会更加困难,势必需要更加细致地优化参数。然而,到目前为止,还未见到有相关文献报道。而这是光化学调价用于真正后处理流程的必经阶段,非常值得研究。从机理研究角度来看,前人的研究已经取得了大量的结果,但是在某些方面还存在争议。很重要的一点是NO-3在其中会起到什么样的作用,因为后处理都是以硝酸为介质,且硝酸浓度还比较高。要解决这一问题,还需要更多的研究结果才行。
光化学方法分离混合体系中金属元素难度较大,目前相关文献报道不多。高放废液中含有大量的Pd、Ru、Rh等铂族金属元素,它们的提取回收有利于高放废液玻璃固化的顺利进行。通过调节反应参数,光化学方法很有希望实现它们的分离回收,但离实际应用也还有很长的路要走。关于Tc的光化学调价,目前研究还很少。但有文献曾报道过利用水溶液辐解产生的水合电子或H原子可以把TcO-4还原成Tc(Ⅵ),Tc(Ⅵ)经歧化反应生成Tc(Ⅳ),Tc(Ⅳ)发生水解最终生成Tc O2·n H2O胶体[82]。采用光化学法有望实现同样的过程,把Tc从高放废液中提取出来,从而解决Tc(Ⅶ)的迁移问题。
[1]叶国安,张虎.核燃料后处理技术发展及其放射化学问题[J].化学进展,2011,23(7):1289-1294.
[2]顾忠茂.我国先进核燃料循环技术发展战略的一些思考[J].核化学与放射化学,2006,28(1):1-10.
[3]胡思思,夏良树,彭安国,等.肼催化还原U(Ⅵ)制备U(Ⅳ)的研究进展[J].核化学与放射化学,2014,36(1):17-23.
[4]Sasaki S,Wada Y,Tomiyasu H.Basic study of photochemistry for application to advanced nuclear fuel cycle technology[J].Progress in Nuclear Energy,1998,32(3-4):403-410.
[5]Enokida Y,Suzuki A.Neptunium valence adjustment through photochemically induced redox reactions at low concentrations[J].J Nucl Sci Technol,1989,26(8):770-776.
[6]Wada Y,Morimoto K,Goibuchi T.Photochemical oxidation of neptunium(Ⅴ)to neptunium(Ⅵ)in nitric-acid solution containing reductants[J].J Nucl Sci Technol,1995,32(10):1018-1026.
[7]Fukasawa T,Kawamura F.Photochemical-reactions of neptunium in nitric-acid solution containing photocatalyst[J].J Nucl Sci Technol,1991,28(1):27-32.
[8]Kessinger G F K,Kyser E A,Almond P M.Reduction of Np(Ⅴ)to Np(Ⅳ)-alternatives to ferrous sulfamate:SRNL-STI-2009-00610[R].South Carolina:Savannah River National Laboratory,2009.
[9]Zielen A J,Sullivan J C,Cohen D.The photochemical reduction and the autoreduction of neptunium(Ⅵ)[J]. J Inorg Nucl Chem,1958,7(4):378-383.
[10]Mazumdar A S,Sivarama C K.A study of nitrate and chloride complexes of plutonium(Ⅵ)by solvent extraction technique using TTA as chelating agent[J].J Inorg Nucl Chem,1965,27(11):2423-2427.
[11]Palei P N,Nemodruk A A,Bezrogova E V,et al. Change of valence states of plutonium in solutions of nitric,hydrochloric,sulfuric,and perchloric acids on irradiation by ultraviolet light[J].Radiokhimiya,1969,11:300.
[12]Bell J T,Friedman H A.Photochemical reactions of aqueous plutonium systems[J].J Inorg Nucl Chem,1976,38(4):831-835.
[13]Friedman H A,Toth L M,Bell J T.Photochemical reactions of aqueous plutonium systems 2[J].J Inorg Nucl Chem,1977,39(1):123-126.
[14]Toth L M,Felker L K.The reduction of Pu(Ⅳ)by photochemically produced U(Ⅳ):ORNL/TM-9958[R]. Tennessee State:Oak Ridge National Laboratory,1986.
[15]Park Y Y,Ikeda Y,Harada M,et al.Oxidation of uranium(Ⅳ)by nitrite ion in dimethyl-sulfoxide-evidence of direct oxygen-transfer mechanism[J]. Chem Lett,1991,1991(8):1329-1332.
[16]Nemodruk A A,Bezrogov E V,Ivanova S A,et al. Photochemical reactions in analytical-chemistry 13:reduction and oxidation of different valent states of neptunium under ultraviolet-light in nitric-acid solutions[J].Zh Anal Khim,1972,27(12):2414-2420.
[17]Friedman H A,Toth L M,Osborne M M.Photochemistry of neptunium in aqueous perchloric-acid solutions[J].J Inorg Nucl Chem,1979,41(9):1339-1345.
[18]Depoorter G L,Rofer-Depoorter C K.The effect of IR laser radiation on the UO2(NO3)2-tributyl phosphate-nitric acid solvent extraction system[J].J Inorg Nucl Chem,1977,39(11):2061-2062.
[19]Gangwer T.Photochemistry relevant to nuclear waste separations:a feasibility study:BNL-50715[R].New York:Brookhaven National Laboratory,1977.
[20]Bell J T,Toth L M.Photo and radiation-chemistry in nuclear-fuel reprocessing[J].Radiochim Acta,1978,25(3-4):225-230.
[21]Tsushima S.Photochemical reduction ofin the presence of alcohol studied by density functional theory calculations[J].Inorg Chem,2009,48(11):4856-4862.
[22]Mc Cleskey T M,Foreman T M,Hallman E E, et al.Approaching zero discharge in uranium reprocessing:photochemical reduction of uranyl[J].Environ Sci Technol,2001,35(3):547-551.
[23]Nagaishi R,Katsumura Y,Ishigure K,et al.Photoreduction of uranyl ion in aqueous solution 1:with ethanol in sulphuric acid solutions[J].J Photochem Photobiol,A,1996,96(1-3):45-50.
[24]Nagaishi R,Katsumura Y,Ishigure K,et al.Photoreduction of the uranyl ion in aqueous solution Ⅱ:alcohols in acid solutions[J].J Photochem Photobiol,A,2002,146(3):157-161.
[25]Wada Y,Wada K,Goibuchi T,et al.Photochemically-induced valency adjustment of plutonium and neptunium in nitric-acid solution using mercury lamp[J].J Nucl Sci Technol,1994,31(7):700-710.
[26]Wada Y,Morimoto K,Goibuchi T,et al.Photochemical valence adjustment of Pu and Np in nitric acid solution for separation and co-extraction[J]. MRS Online Proceedings,1994,353:1347-1354.
[27]Wada Y,Morimoto K,Tomiyasu H.Separation and coextraction of Pu and Np by solvent extraction using 30%TBP/n-dodecane after and during photochemical valence adjusting[J].Radiochim Acta,1996,72(4):195-204.
[28]Shilov V P,Yusov A B.Photochemical reactions of plutonium ions in perchloric acid solutions[J].Radiochemistry,2001,43(4):364-370.
[29]Nishida D,Kusaba M,Yatsuhashi T,et al.Reduction of Eu3+to Eu2+by an intense femtosecond laser pulse in solution[J].Chem Phys Lett,2008,465:238-240.
[30]Harada M,Inada Y.In situ time-resolved XAFS studies of metal particle formation by photoreduction in polymer solutions[J].Langmuir,2009,25(11):6049-6061.
[31]Harada M,Tamura N,Takenaka M.Nucleation and growth of metal nanoparticles during photoreduction using in situ time-resolved SAXS analysis[J].J Phys Chem C,2011,115(29):14081-14092.
[32]Li H X,Lin M Z,Hou J Z.Electrophoretic deposition of ligand-stabilized silver nanoparticles synthesized by the process of photochemical reduction[J]. J Cryst Growth,2000,212(1-2):222-226.
[33]Kusaba M,Tsunawaki Y,Nakashima N.Formation of Cd particles by UV laser irradiation[J]. Thin Solid Films,2008,517(4):1500-1502.
[34]Wang S,Gao Y L,Wang T,et al.Preparation andphotocatalytic performance of Pt-TiNT@@SiO2by UV photoreduction[J].Chin J Catal,2011,32(9):1513-1518.
[35]Balzani V C V.Photochemistry of co-ordination compounds[M].London:Academic Press,1970.
[36]Burrows H D,Kemp T J.The photochemistry of the uranyl ion[J].Chem Soc Rev,1974,3(2):139-165.
[37]Kannan S,Vaughn A E,Weis E M,et al.Anhydrous photochemical uranyl(Ⅵ)reduction:unprecedented retention of equatorial coordination accompanying reversible axial oxo/alkoxide exchange[J].J Am Chem Soc,2006,128(43):14024-14025.
[38]Latimer W M.Oxidation potentials[M].New York:Prentice-Hall Inc,1952.
[39]Baird C P,Kemp T J.Luminescence,spectroscopy,lifetimes and quenching mechanisms of excited states of uranyl and other actinide ions[J].Prog React Kinet,1997,22(2):87-139.
[40]Volman D H,Seed J R.Photochemistry of uranyl oxalate[J].J Am Chem Soc,1964,86(23):5095-5098.
[41]Porter K,Volman D H.Uranyl oxalate actinometer for microphotochemistry[J].J Am Chem Soc,1962,84(10):2011-2012.
[42]Heidt L J,Tregay G W,Middleto F.Influence of p H upon photolysis of uranyl oxalate actinometer system[J].J Phys Chem,1970,74(9):1876-1882.
[43]McBrady J J,Livingston R.The formation of tetravalent uranium during the uranyl-sensitized photochemical decomposition of oxalic acid[J].J Phys Chem,1946,50(3):176-190.
[44]Brits A G,Vaneldik R,Vandenberg J A.Photolysis of uranyl oxalate system 3:photochemical behavior,kinetics and mechanism in aqueous-solution[J].J Inorg Nucl Chem,1977,39(7):1195-1199.
[45]Brits A G,Vaneldik R,Vandenberg J A.Photolysis of uranyl formic acid formate system in acidic aqueous-solution[J].Inorg Chim Acta,1978,30(1):17-22.
[46]Heckler G E,Jensen R,Jensen C,et al.Uranyl sensitized photodecomposition of organic acids in solution[J].J Phys Chem,1963,67(1):1-6.
[47]Greatore D,Stone T J,Kemp T J,et al.Electronspin resonance studies of photooxidation by metalions in rigid media at low-temperatures 4:survey of photooxidation by uranyl ion[J].J Chem Soc, Faraday Trans,1972,68(11):2059-2076.
[48]Sakuraba S,Matsushi R.Photochemical reactions of uranyl ions with organic compounds 2:mechanism of photo-oxidation of alcohols by uranyl ions[J].Bull Chem Soc Jpn,1970,43(8):2359-2363.
[49]Sakuraba S,Matsushi R.Photochemical reactions of uranyl ions with organic compounds 4:uranyl fluorescence quenching by aliphatic alcohols[J]. Bull Chem Soc Jpn,1971,44(11):2915-2918.
[50]Matsushi R,Sakuraba S.Substituent effects on photochemical reaction rates of uranyl-alcohol system[J].J Am Chem Soc,1971,93(21):5421-5423.
[51]Matsushi R.Mechanism of quenching of uranyl fluorescence by organic compounds[J].J Am Chem Soc,1972,94(17):6010-6016.
[52]Hill R J,Kemp T J,Allen D M,et al.Absorptionspectrum,lifetime and photoreactivity towards alcohols of excited-state of aqueous uranyl-ion()[J].J Chem Soc,Faraday Trans,1974,70(5):847-857.
[53]Ledwith A,Russell P J,Sutcliff L H.Alkoxy radical intermediates in thermal and photochemical oxidation of alcohols[J].Proc R Soc Lond A-Math Phys Sci,1973,332(1589):151-166.
[54]Salomone V N,Meichtry J M,Schinelli G,et al. Photochemical reduction of U(Ⅵ)in aqueous solution in the presence of 2-propanol[J].J Photochem Photobiol,A,2014,277:19-26.
[55]Gonzalez M G,Oliveros E,Worner M,et al.Vacuum-ultraviolet photolysis of aqueous reaction systems[J].J Photochem Photobiol C-Photochem Rev,2004,5(3):225-246.
[56]段云富,张先业,周志宏,等.Pu(Ⅲ)-Fe(Ⅱ)-N2H+5-HNO3溶液中的光化反应[J].核化学与放射化学,1987,9(4):200-206.
[57]Toth L M,Friedman H A.The photochemistry of neptunium in aqueous nitric-acid solutions[J].Radiochim Acta,1980,27(3):173-176.
[58]Galkin B Y,Krupitskii S V,Lazarev L N,et al.Photooxidation of neptunium in nitric-acid solutions[J]. Soviet Radiochemistry,1985,27(4):455-458.
[59]Shilov V P,Yusov A B.Mechanisms of photochemical reactions of neptunium ions in nitric acid solutions[J].Radiochemistry,1996,38(5):398-401.
[60]沈东,范显华,苏锡光,等.锝在磁黄铁矿上的吸附行为和机理的研究[J].核化学与放射化学,2001,23(2):72-78.
[61]Lieser K H,Bauscher C.Technetium in the hydrosphere and in the geosphere 1:chemistry of technetium and iron in natural-waters and influence of the redox potential on the sorption of technetium[J].Radiochim Acta,1987,42(4):205-213.
[62]Burton-Pye B P,Radivojevic I,McGregor D,et al. Photoreduction of99Tc pertechnetate by nanometersized metal oxides:new strategies for formation and sequestration of low-valent technetium[J].J Am Chem Soc,2011,133(46):18802-18815.
[63]Friedman H A,Toth L M.Photochemically induced reduction of trace Np(Ⅵ)in U(Ⅵ)-HNO3solutions[J].J Inorg Nucl Chem,1981,43(7):1611-1613.
[64]Uchiyama G,Kihara T,Hotoku S,et al.Development of a new neptunium separation process by using photochemical reduction in nuclear fuel reprocessing[J].Radiochim Acta,1998,81(1):29-32.
[65]Donohue T.Photochemical separation of Europium from lanthanide mixtures in aqueous-solution[J].J Chem Phys,1977,67(11):5402-5404.
[66]Haas Y,Stein G,Tenne R.Photochemistry of solutions of Eu(Ⅲ)and Eu(Ⅱ)[J].Isr J Chem,1972,10(2):529-536.
[67]Nishida D,Yamade E,Kusaba M,et al.Reduction of Sm3+to Sm2+by an intense femtosecond laser pulse in solution[J].J Phys Chem A,2010,114(18):5648-5654.
[68]Tsushima S,Nagasaki S,Suzuki A.Photochemical separation and co-precipitation of lanthanides in nitric acid solution[J].J Photochem Photobiol,A,1997,106(1-3):57-60.
[69]Tsushima S,Nagasaki S,Suzuki A.Separation of lanthanides and oxidation of Am in nitric acid solution by photolysis[J].J Nucl Sci Technol,1995,32(2):154-156.
[70]Fredrickson J K,Zachara J M,Kennedy D W,et al.Reduction of TcO-4by sediment-associated biogenic Fe(Ⅱ)[J].Geochim Cosmochim Acta,2004,68(15):3171-3187.
[71]Song K,Cha H,Lee J,et al.Extraction of palladium metal from aqueous solution of palladium chloride by laser-induced photochemistry[J].Microchem J,2001,68(2-3):121-126.
[72]Harada M,Takahashi S.Synthesis of ruthenium particles by photoreduction in polymer solutions[J]. J Colloid Interface Sci,2008,325(1):1-6.
[73]Ohtaki M,Toshima N.Photoreduction of rhodium(Ⅲ)ions in water with ultraviolet-light aiming to prepare the dispersions of ultrafine particles[J].Chem Lett,1990,(4):489-492.
[74]张昊,谭欣,赵林.废水中重金属离子的光催化还原研究进展[J].天津理工学院学报,2004,20(03):28-32.
[75]刘国光,丁雪军,张学治,等.光催化氧化技术的研究现状及发展趋势[J].环境污染治理技术与设备,2003,4(8):65-69.
[76]Kriek R J,Engelbrecht W J,Cruywagen J J.Separation and recovery of some platinum-group metals(PGMs)by means of selective photocatalytic reduction[J].J S Afr Inst Min Metall,1995,95(2):75-81.
[77]Borgarello E,Serpone N,Emo G,et al.Light-induced reduction of rhodium(Ⅲ)and palladium(Ⅱ)on titanium dioxide dispersions and the selective photochemical separation and recovery of gold(Ⅲ),platinum(Ⅳ),and rhodium(Ⅲ)in chloride media[J].Inorg Chem,1986,25(25):4499-4503.
[78]Ikeda Y,Yasuike Y,Nishimura K,et al.Kinetic study on dissolution of UO2powders in nitric acid[J]. J Nucl Mater,1995,224(3):266-272.
[79]Ikeda Y,Yasuike Y,Takashima Y,et al.17O NMR study on dissolution reaction of UO2in nitric acid-mechanism of electron-transfer[J].J Nucl Sci Technol,1993,30(9):962-964.
[80]Kim E H,Hwang D S,Yoo J H.Dissolution mechanism of UO2in nitric acid solution by photochemical reaction[J].J Radioanal Nucl Chem,2000,245(3):567-570.
[81]Kim E H,Choung W M,Kim Y K,et al.A photoinduced dissolution of UO2sintered pellets in a simulated solution[J].Korean J Chem Eng,2000,17(2):217-222.
[82]Zakir M,Sekine T,Takayama T,et al.Technetium(Ⅳ)oxide colloids and the precursor produced by bremsstrahlung irradiation of aqueous pertechnetate solution[J].J Nucl Radiochem Sci,2005,6(3):243-247.
photochemical Adjustment Valence of Actinides and precious Metal Elements in Reprocessing process
LI Fu-hai1,DING Zuo-ming1,LIN Ming-zhang1,2,*,HE Hui3,YE Guo-an3
1.School of Nuclear Science and Technology,University of Science and Technology of China,Hefei 230027,China;2.Institute of Nuclear Energy Safety Technology,Chinese Academy of Science,Hefei 230031,China;3.China Institute of Atomic Energy,P.O.Box 275(26),Beijing 102413,China
The PUREX process is the only reprocessing process applied on an industrial scale in the world.One of the key technologies of this process is to adjust the valence of plutonium(Pu),neptunium(Np)and technetium(Tc).Traditionally the valence of these elements should be adjusted by chemical methods.Chemical methods,however,might meet difficulties such as the valence adjustment of Np in nitric acid solution.Compared with the chemical methods,photochemical methods offer the remarkable advantages such as simpler steps,less secondary wastes,easier remote control techniques and low requirements for radioactive resistance of materials,etc.The combination of photochemical methods and solvent extraction offers a potential for the separation of metal elements,and it is also possible to separateand recover metal elements by using photochemical methods to reduce metal ions to atoms. In addition,exposure of UO2under UV light can significantly promote the dissolution rate of UO2at room temperature in nitric acid solution.
photochemistry;valence adjustment;separation;dissolution of UO2
O644.14
A
0253-9950(2016)02-0065-14
10.7538/hhx.2016.38.02.0065
2015-01-09;
2015-06-24
国家自然科学基金资助项目(21377122)
李富海(1991—),男,河南濮阳人,硕士研究生,核科学与技术专业
*通信联系人:林铭章(1965—),男,福建泉州人,博士,教授,博士生导师,从事辐射化学与放射化学研究,E-mail:gelin@ustc.edu.cn
