青海湖西岸镭同位素的解吸和扩散特征*
2016-10-12孔凡翠沙占江杜金洲苏维刚胡菊芳王求贵马玉军翟玉乐马海英
孔凡翠,沙占江,,杜金洲,苏维刚,胡菊芳,5, 王求贵,5,马玉军,翟玉乐,王 转,马海英
(1:中国科学院青海盐湖研究所,青海省盐湖地质与环境重点实验室,西宁 810008) (2:青海师范大学,青海省自然地理与环境过程重点实验室,西宁 810008) (3:华东师范大学,河口海岸国家重点实验室,上海 200062) (4:青海省地震局预报中心,西宁 810001) (5:中国科学院大学,北京 100049) (6:青海省德令哈市国土资源局,德令哈 817000)
青海湖西岸镭同位素的解吸和扩散特征*
孔凡翠1,沙占江1,2**,杜金洲3,苏维刚4,胡菊芳1,5, 王求贵1,5,马玉军2,翟玉乐2,王转2,马海英6
(1:中国科学院青海盐湖研究所,青海省盐湖地质与环境重点实验室,西宁 810008) (2:青海师范大学,青海省自然地理与环境过程重点实验室,西宁 810008) (3:华东师范大学,河口海岸国家重点实验室,上海 200062) (4:青海省地震局预报中心,西宁 810001) (5:中国科学院大学,北京 100049) (6:青海省德令哈市国土资源局,德令哈 817000)
对青海湖布哈河河口悬浮颗粒物、底部沉积物和青海湖湖底沉积物中的镭(Ra)同位素进行不同盐度和pH值的解吸实验以及扩散实验,得到不同盐度湖水(2.8‰、5.8‰、8.8‰、11.8‰和14.8‰)对悬浮颗粒物中镭的解吸活度,和不同时间段沉积物中镭同位素的扩散速率,探讨盐度、pH值与颗粒物中镭同位素解吸的关系. 结果表明,224Ra的解吸活度均高于226Ra和228Ra的解吸活度;在盐度为12‰附近时布哈河河口悬浮颗粒物中223Ra、226Ra和228Ra的解吸程度达到最大值,当盐度<9‰时,226Ra解吸活度大于228Ra,当盐度>9‰时,228Ra的解吸活度大于226Ra,这可能与当地岩石中富铀矿有关. 河流沉积物226Ra和228Ra的扩散速率分别是0.039和0.290dpm/(m2·h);湖底沉积物226Ra和228Ra的扩散速率分别为0.018和0.092dpm/(m2·h),湖底沉积物扩散速率小于河流沉积物扩散速率.
镭同位素;解吸;扩散;布哈河口;青海湖
(3: State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai 200062, P.R.China)
(4: Qinghai Seismological Bureau Forecast Center, Xining 810001, P.R.China)
(5: University of Chinese Academy of Sciences, Beijing 100049, P.R.China)
(6: The Land and Resources Bureau of Delingha City, Delingha 817000, P.R.China)
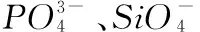
悬浮颗粒物上镭同位素的解吸过程发生较快,在几秒到几小时的时间内就完成,通常在淡咸水交汇的几分钟内就能达到解吸量的30%~50%[5]. 辽河口水体中的224Ra和223Ra随盐度变化呈不保守现象,研究认为盐度为10‰左右的海区镭同位素高峰值可能是悬浮颗粒物解吸造成的[36]. 水体pH值影响颗粒物的表面净电荷量,在低pH值的水体环境中,二价镭离子形成表面络合物的能力较低,进行解吸的能力较强[37-38].Gonneea等研究认为,在水体pH值从8降低至5时,粗砂上的镭较易发生解吸[26]. 在青海湖研究区域,布哈河河水的盐度变化范围为0.2‰~0.6‰,青海湖湖水盐度变化范围为10.10‰~14.71‰,从河水到湖水盐度变化比较大,盐度可能是青海湖镭解吸的一个很重要的影响因素. 但是前人研究内容多为研究区盐度或者pH值对颗粒物解吸活度的影响,或者是海底沉积物扩散镭的活度,在一个研究区同时进行镭同位素解吸和扩散实验的报道较少. 而且前人对于沉积物中镭的吸附-解吸行为的研究都是在沿海地区,而在高原内陆咸水湖的研究还未见报道.
本文为了更好地研究镭同位素的来源,在已知河流溶解态、地下水、湖泊表层水中镭同位素活度的基础上,拟通过开展不同盐度和pH值条件下,青海湖布哈河河口悬浮颗粒物中镭同位素的解吸实验,河流底部沉积物以及湖底沉积物中镭同位素的扩散活度实验,推算高原内陆咸水湖泊周围河流悬浮颗粒物中镭同位素的解吸与盐度、pH值之间的关系和扩散活度值特征,通过对沉积物中镭的解吸量和扩散量进行研究,从而为辨别青海湖水体中镭同位素的来源、评估入湖水体停留时间、示踪沿岸地下水的排放通量推算研究提供科学依据.
1 理论模型及实验过程
1.1 解吸实验的理论模型及过程
在青海湖流域河流入湖处,布哈河河水的盐度均小于1‰,而青海湖的盐度为14‰左右,在咸水和淡水混合区,在影响镭的吸附-解吸行为的众多因素中,水体盐度的影响对镭的吸附-解吸行为最为重要[17-18,26],我们就水体盐度梯度下镭在固-液两相的分配行为进行讨论,以期为辨别镭同位素来源、示踪沿岸地下水排放通量、评估水体交换时间以及估算沉积物-水界面交换通量等研究提供必要的定量依据.
Webster等曾就水体盐度梯度下镭在颗粒物上的解吸行为,建立了理论依据相对充分的数学模型,并进行室内解吸实验以验证模型的可靠性[18]. 在该理论模型中,认为受水体盐度变化的影响,水体中镭的增加可通过式(1)加以描述:
(1)
其中,S为水体盐度值,表示占标准海水的千分比,单位为‰;Aw为颗粒物解吸进入水体中镭的比活度,单位为dpm/g;A0和a/b为颗粒物的解吸参数,两者数值大小取决于颗粒物的本身特性;A0的物理意义为颗粒物尚未发生解吸时,单位面积的颗粒物上“可交换态”镭的活度量,单位为dpm/g;a/b是公式推导过程中,由Na+和Ra2+的吸附和解吸常数、颗粒物表面积、颗粒物表面能够吸附Na+和Ra2+最大个数等参数组成的函数,无量纲;R为解吸实验中水体体积与颗粒物体积的比值,即固-液比的倒数.
若根据式(1)指导解吸实验,只需设定R,并测量已知盐度系列下的水体镭浓度Aw,即可根据最小二乘法确定颗粒物解吸参数a/b和A0的数值,此为第一种解吸实验方案,通过不同盐度条件下颗粒物中镭的解吸分析镭同位素在固-液两相的分配行为.

(2)
式中,Q为等比数列的公比数. 第2次沉积物解吸后水体镭浓度增幅需满足质量平衡,即:
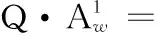
(3)
由此确定:
(4)
根据递推法则,第n次解吸后水体镭浓度应等于该等比数列的前n项之和,即:
(5)
根据以上理论推算,颗粒物若在同一特定盐度的水体中多次解吸,则根据每次解吸后水体镭浓度增加趋势,结合最小二乘法,同样可以拟合出颗粒物解吸参数a/b和A0的数值,此为第2种解吸实验方案,即用同一个盐度对悬浮颗粒物进行多次淋洗分析镭同位素在固-液两相的分配行为.
本研究结合以上两种解吸实验方案思路,即由第2种方案确定布哈河悬浮颗粒物中镭是否发生解吸,然后由第一种方案确定镭同位素在固-液中的分配行为.
首先用青海湖湖水对布哈河河口悬浮颗粒物进行淋洗,如果能从悬浮颗粒物上淋洗出镭同位素,那说明青海湖湖水会对河流悬浮颗粒物中的镭发生解吸,否则将不会发生解吸. 淋洗实验过程:选择盐度为14.8‰ 的青海湖湖水对悬浮颗粒物进行多次淋洗实验,具体操作步骤如下:
1) 对采集的青海湖湖水用锰纤维进行过滤吸附其中含有的镭,形成无镭水,用圆柱形水桶取25L;
2)将15g河流颗粒物置于水桶中搅拌2h,静止沉淀1h;
3)过滤上清液富集悬浮颗粒物解吸出来的镭;
4)富集后立刻用RaDeCC器测试224Ra活度值;
5)检测富集镭后的滤液盐度以确保未发生明显变化,倒入水桶中再次搅拌2h进行淋洗,如此反复7次,每次淋洗后的滤液均采用同一根锰纤维柱富集,并立即测量224Ra活度值,直到锰纤维柱的224Ra活度不再增加为止.
8次淋洗实验在24h内连续快速完成,并测试224Ra的活度,以避免224Ra的衰减成为干扰因素.
随着不断对布哈河中悬浮颗粒物进行淋洗,溶解态224Ra的活度不断增加,但是当淋洗到第7次时,溶解态224Ra的活度呈现稳定趋势,达到最大解吸活度(3.592dpm/g),表明河流悬浮颗粒物中的镭被不断解吸到河流水体中,最后趋于稳定(图1).
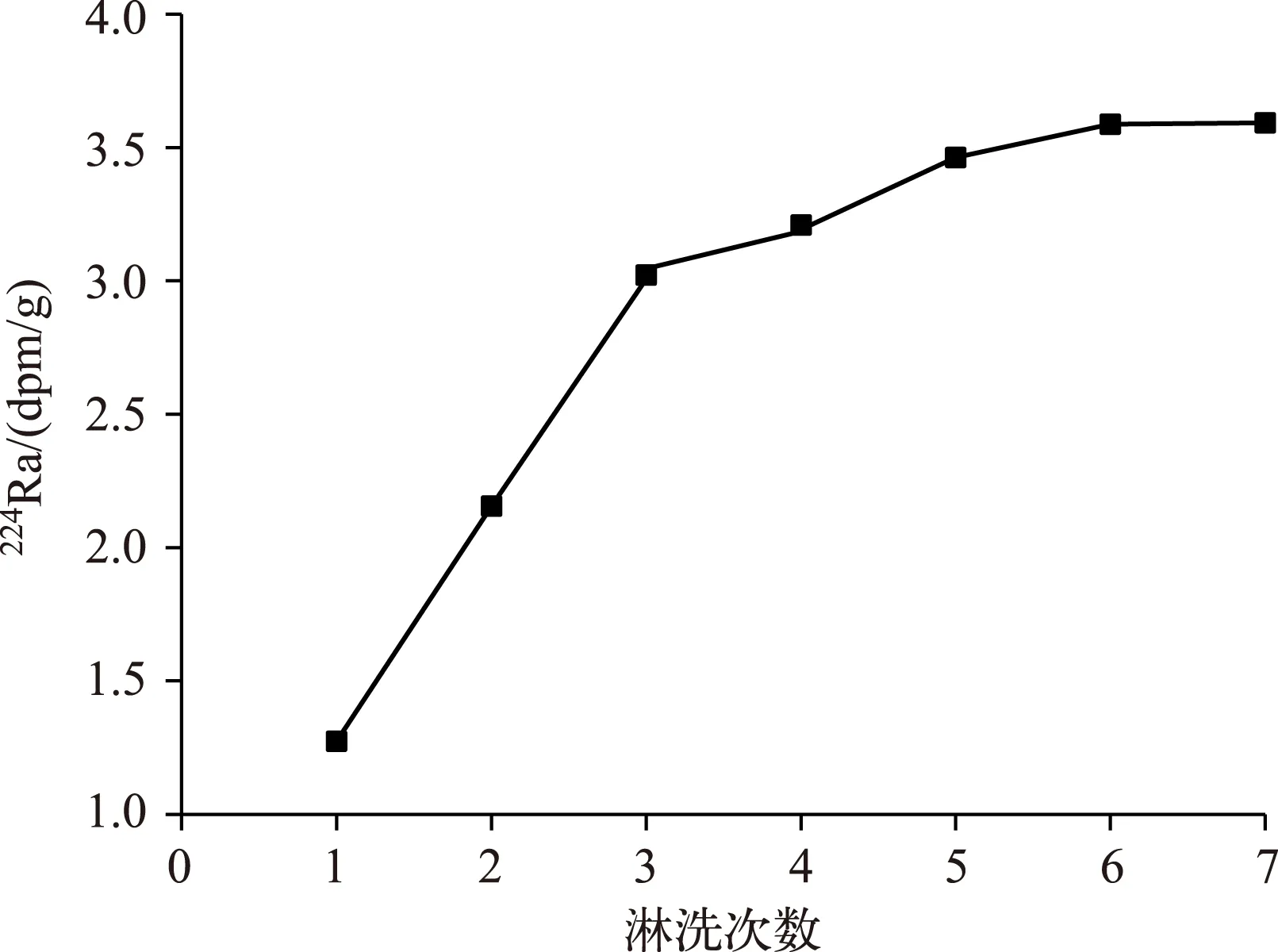
图1 224Ra淋洗实验结果Fig.1 The result of 224Ra leaching experiment
由淋洗实验可知,河流悬浮颗粒物中的镭遇到青海湖湖水会生解吸,因此可以进行河流悬浮颗粒物中镭的解吸实验,不同盐度解吸操作过程为:
1)配置无镭湖水. 经测定青海湖湖水盐度最大值为14.8‰,取足量盐度为14.8‰左右的湖水,流过装有MnO2纤维柱子制成无镭湖水,通过往无镭湖水中加去离子水配置成盐度分别为11.8‰、8.8‰、5.8‰、2.8‰、0.1‰(盐度为0无法配制)的无镭水,每份体积为25L;
2)取6份干的河流悬浮颗粒物,每份10g置于上述盐水中,利用搅拌机连续搅拌2h后静止沉淀,搅拌强度以使颗粒物恰好悬浮为宜,静止沉淀1h;
3)用0.45μm滤膜过滤上清液,将收集的滤液按照前面的方法用虹吸方式让沉积物上覆水缓慢流过MnO2纤维柱来富集镭,立即测量224Ra活度值;
4)测完224Ra活度值的锰纤维进行共沉淀流程获得Ba(Ra)SO4固体沉淀,将沉淀放入γ谱仪测试226Ra和228Ra的活度值.
1.2 底部沉积物中镭同位素的扩散实验过程
自然界存在的镭同位素由沉积物中母体U-Th衰变而来. 河口表层沉积物中U-Th衰变产生的镭,通常使得沉积物间隙水中镭活度大于河口水中的镭活度,在活度差作用下,间隙水中的镭扩散到河口水体中,成为河口水中镭的来源之一[39]. 分别采集湖底沉积物和布哈河河口沉积物进行扩散培养实验. 把采集的表层沉积物分别放置到直径为25和36cm的敞口塑料桶中,厚度5~10cm,对沉积物进行不同时段的扩散培养实验. 为了使其充分稳定下来,在实验之前将该样品放置2个月,因为在取样和搬运过程中会对样品产生一定扰动,对测量结果有影响[40].
对湖底沉积物样品和河流沉积物样品分别进行0.5、l、2、4、7、12和28d的扩散培养,具体实验过程为[39]:在湖底沉积物扩散实验中:沿着桶壁缓慢加入盐度为14.8‰的无镭湖水20L,尽量不扰动沉积物,放置培养. 当达到培养时间后对水体中镭进行富集和测量. 之后,再加入无镭湖水按照上述步骤进行下一个时间段的扩散培养;在河流沉积物扩散实验中:每个时段开始时,沿桶壁缓慢加入盐度为0.2‰的无镭河口水25L,沉积物间隙水中的镭会逐渐扩散到该上覆水体中,每个时段结束时,对水体中的镭进行富集并测量其活度,然后再加入新的无镭河口水进行下一个时段的扩散培养,在每次加入无镭河口水和富集水体中的镭时,都尽量减少对沉积物的扰动.
2 样品采集及测试方法
布哈河是青海湖湖区最大的地表径流,位于青海湖西北部,河长280km,集流面积14300km2,约占青海湖流域面积的一半,水系呈不对称的羽状分布,年平均径流量7.85×108m3,年总输沙量为3.55×105t,占总量的70%以上[41]. 青海湖湖底沉积物扩散实验的沉积物是2012年7月在站位QH1、QH2、QH3(图2)用沉积物采样器采集的3个沉积物柱子,共2kg. 2014年8月在布哈河选择河水盐度为0.1‰(未找到盐度为0的河水端元)的站位R1过滤河水采集河流悬浮颗粒物5kg,在站位R2和R3(图2)采集河口区河流底部沉积物共10kg,分别用于河流悬浮颗粒物的解吸和河流底部沉积物的扩散实验,其中站位R2、R3河水的盐度分别为0.2‰、0.3‰,pH值分别为8.19、8.59.
所有样品均通过锰纤维富集,短半衰期223Ra和224Ra采用同步延时计数器(RaDeCC)测量,该仪器为美国ScientificComputerInstruments公司生产的四通道同步延时计数器. 长半衰期226Ra和228Ra放射性活度的测试用的是美国ORTEC公司生产的高纯锗γ谱仪,探测器为GMX45P4型,所有实验和测试均在中国科学院青海盐湖研究所完成.
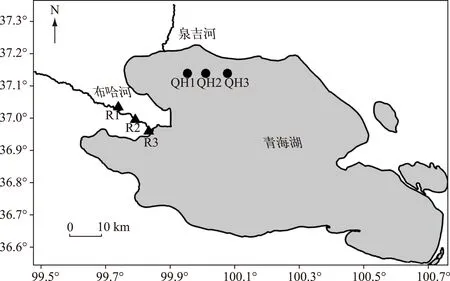
图2 布哈河河口悬浮颗粒物、底部沉积物和青海湖湖底沉积物采样点位置Fig.2 Location of sampling sites of suspended particles and sediments of Buha River estuary and sediments of Lake Qinghai
3 结果与讨论
3.1 悬浮颗粒物中镭同位素的解吸特征
盐度是影响镭解吸的重要因素,在一般封闭体系中,沉积物中镭与其母体钍处于平衡状态,当遇到水体盐度发生变化时,如河流搬运悬浮颗粒物至河口盐度梯度区,或海水倒灌入侵海岸含水层,形成地下河口盐度梯度区,沉积物上的镭就会与海水中常量阳离子如Na+等发生离子交换而进入到水体,并随之迁移,形成沉积物中镭相对于母体钍亏损的现象[18]. 布哈河河口悬浮颗粒物不同盐度(0、5.8‰、8.8‰、11.8‰和14.8‰)所对应的悬浮颗粒物中224Ra的解吸活度分别为0.43、0.57、0.59、0.37和0.32dpm/g,盐度为2.8‰时没有检测到224Ra的活度;盐度为8.8‰、11.8‰和14.8‰所对应的223Ra的解吸活度分别为0.025、0.069和0.036dpm/g,盐度为2.8‰和5.8‰时没有检测到223Ra的活度;各盐度所对应的226Ra的解吸活度分别为0.29、0.15、0.18、0.16、0.21和0.13dpm/g;各盐度所对应的228Ra的解吸活度分别为0.14、0.11、0.14、0.18、0.23和0.17dpm/g. 布哈河悬浮颗粒物中224Ra的解吸活度均高于对应的226Ra和228Ra的解吸活度,这可能与224Ra再生速率快有关(表1). 布哈河河口悬浮颗粒物中226Ra的解吸活度远小于海南东部八门湾悬浮颗粒物226Ra的解吸活度3.93dpm/g[42],大于胶州湾沉积物226Ra的解吸活度(0.07~0.20dpm/g)[29],与九江河口区悬浮颗粒物中226Ra的解吸活度(0.13~0.25dpm/g)相接近[39](表1). 说明本次实验获得的解吸数据是比较可信的.
Gonneea等发现当盐度升高时,镭的解吸量具有增大的特点[26]. 随着盐度的逐渐增加,Ra同位素的解吸活度先呈增加趋势,说明盐度越高,悬浮颗粒上解吸下来的Ra越多. 从理论上分析, 镭的解吸量不可能无限制地增加,随着盐度的继续增加,224Ra的解吸活度表现为降低趋势(图3a). 当盐度<9‰时,226Ra的解吸活度大于228Ra,当盐度>9‰时,228Ra的解吸活度大于226Ra(图4). 在盐度为12‰时,226Ra和228Ra的解吸活度达到了最大. 当盐度<4‰时,Ra的解吸量增加较为缓慢,当4‰<盐度<12‰时,Ra的解吸量增加较迅速,可是当盐度>12‰时,Ra的解吸量开始下降(图3a). 而其他地方的河流悬浮颗粒物Ra的解吸量随盐度的变化有所不同,在九江河口区226Ra解吸活度最大值发生在盐度为9‰时,长江口解吸活度最大值发生在盐度为9‰时,密西西比河解吸活度最大值发生在盐度为5‰时[43-44],Cable等研究了海底沉积物Ra的解吸,认为在盐度为18‰左右Ra的解吸最强烈[25]. 但是都有一个相同的规律,就是随着盐度的升高,镭同位素的解吸量先增大,达到一个最大值后开始减小.
Kiro等对死海沿岸含水层Ra的分布模式研究发现, 短半衰期Ra同位素的活度随着盐度的降低而降
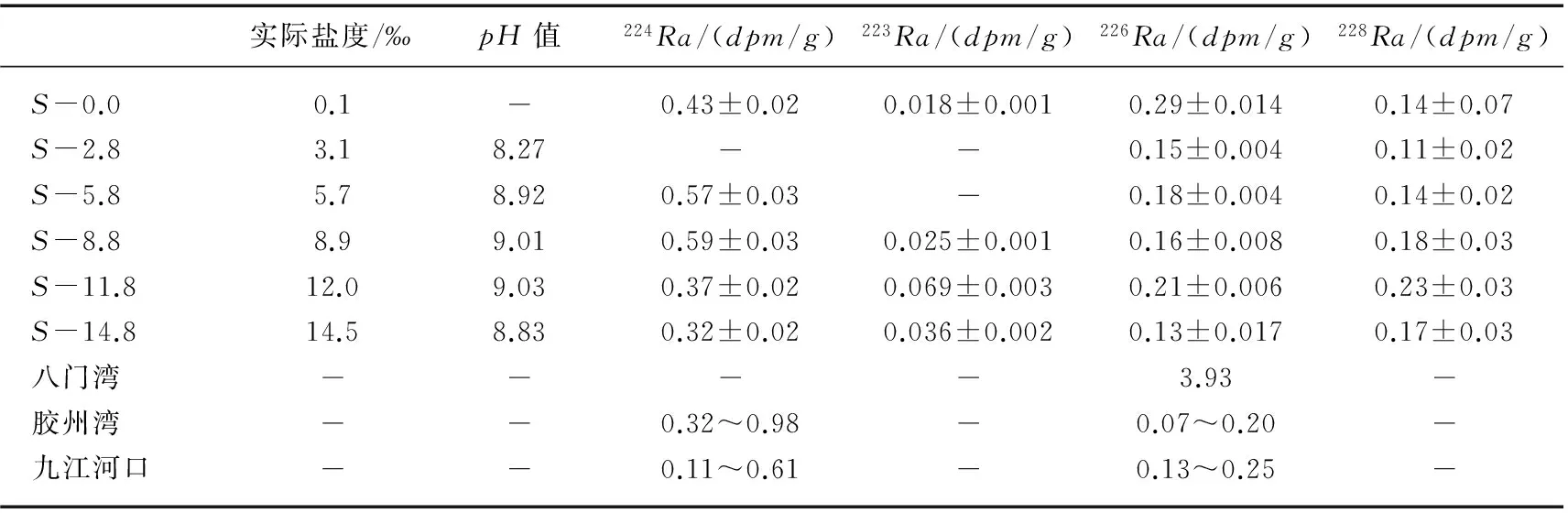
表1 布哈河河口悬浮颗粒物Ra的解吸量Tab.1 Desorption Radium activity of suspended particles in the Buha River estuary
低,是由于盐度对Ra同位素解吸有影响[45]. 辽河口水体中的224Ra和223Ra随盐度变化呈不保守现象,盐度为10‰左右的海区Ra同位素高峰值可能是悬浮颗粒物解吸造成的[36].Moore对Chesapeake湾的Ra同位素研究时,发现该湾内水体中高226Ra和228Ra活度值主要来源于海底表层沉积物的扩散和河口颗粒物的解吸[46]. 对Amazon河口226Ra和228Ra行为的研究发现,来自河口及附近陆架沉积物的Ra扩散提供了Amazon体系总Ra的1/3,其余2/3来自河水溶解态及河流悬浮颗粒的解吸[47].Astwood通过一系列解吸实验得出亚马逊河口区有40%的224Ra来自悬浮颗粒的解吸[48].Krest等对Mississippi河和Atchafalaya河与海水混合过程进行研究,发现悬浮颗粒物上有大约50%的226Ra和228Ra发生解吸,其解吸活度达到18.0dpm/100L[22]. 苏妮等研究海南东部瀉湖以及河口海底地下水发现,万泉河口区226Ra的最大解活度为3.81dpm/100L,八门湾地区226Ra的最大解吸活度为3.42dpm/100L,解吸态的Ra的活度不可忽略[16,49]. 布哈河悬浮颗粒物226Ra的最大解吸量达0.21dpm/g,而河流平均含沙量为0.41g/L,那么河流悬浮颗粒物中226Ra的最大解吸活度为8.4dpm/100L,布哈河水体中溶解态226Ra的活度为8.30±0.24~21.63±0.37dpm/100L,河流悬浮颗粒物中226Ra的解吸活度占到了河流水体中226Ra活度的38.83%. 由此可见,布哈河河口悬浮颗粒物解吸的Ra是青海湖水体中Ra的一项不可忽略的来源,而且在青海湖表层水体中Ra同位素的活度分布呈现出由沿岸向湖中心跳跃式的减少[50],也可能是受到颗粒物解吸的影响.
从布哈河河口悬浮颗粒物中Ra同位素解吸量与pH值的关系可以看到,随着pH值的增加,226Ra和228Ra的解吸量呈逐渐增高趋势,趋势与盐度对其解吸影响一致(图3b). 实验结果显示,pH值对悬浮颗粒物中224Ra、223Ra、226Ra、228Ra解吸的影响不同. 在pH值小于8.8的情况下没检测到223Ra和224Ra的解吸量,对于223Ra、224Ra的解吸量与pH值的关系不好解释.226Ra和228Ra解吸量随着pH值的升高表现出明显的增加趋势.Beck等在美国弗吉尼亚州GloucesterPoint沙滩实验的研究表明,孔隙水的pH值对Ra解吸的影响可以与盐度对Ra解吸的影响相提并论,当pH值在2~10范围内时,Ra的固液分配系数可以变化1~2个数量级,当pH值的突跃范围为5~8时,pH值微小变动将引起Ra的固液分配系数发生10倍左右的变化[27]. 而在青海湖研究区,布哈河河水的pH值变化范围为8.19~8.59,青海湖湖水pH值变化范围为9.02~9.30,从河水到湖水pH值较小的范围内Ra的解吸量与盐度为2‰~14.8‰时Ra的解吸量一样. 因此,从本实验也可以看出,pH值的变化对青海湖Ra解吸的影响较大,并且比盐度对Ra解吸的影响更明显(图4).
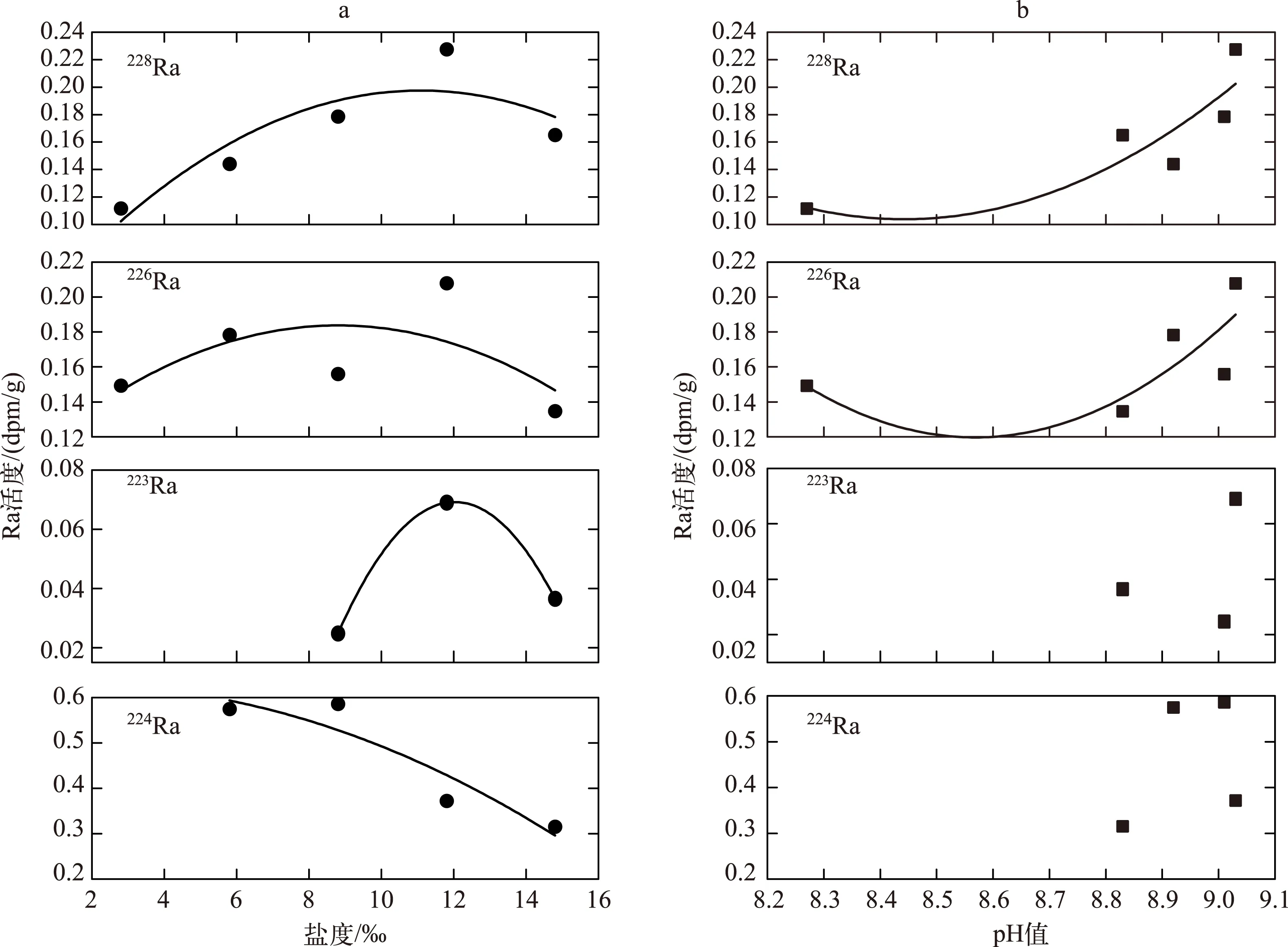
图3 布哈河河口悬浮颗粒物中镭解吸量与盐度(a)和pH值(b)的关系Fig.3 Relationships between desorption Radium activity of suspended particles and salinity (a) and pH values (b) in Buha River estuary
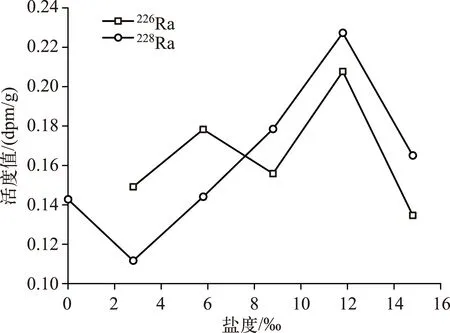
图4 布哈河河口盐度与226Ra、228Ra解吸量的关系Fig.4 Relationship between salinity and distribution of solid-liquid in Buha River estuary
3.2 河流底部沉积物和湖底沉积物中镭同位素的扩散特征
不同培养时间段的河流和湖底沉积物Ra扩散量见表2,由于本次实验检测的224Ra活度是总的224Ra,未检测并去掉母体中224Ra的活度值,计算出的224Ra活度值不准确,在此不再对224Ra的扩散速率进行考虑. 从培养时间与扩散通量的关系可以看出,扩散开始时河流中226Ra和228Ra的活度比较高,随着扩散时间的增加表现出降低的趋势,扩散96h后,又呈现增加的趋势,最后趋于稳定(图5). 这种现象可能是由于Ra同位素的解吸反应在几秒到几小时内就可以完成,当培养时间较短时,Ra的活度普遍偏高,且没有规律可循,因此,培养刚开始时镭的活度主要来自沉积物中原有镭同位素的解吸,来自扩散的Ra不明显. 过一段时间后,沉积物间隙水中Ra活度与新加入的无Ra河口水Ra活度之差达到最大,Ra活度梯度也达到最大,这就会促使分子扩散作用加剧,沉积物孔隙水中Ra同位素的扩散作用开始增强,孔隙水中Ra同位素浓度开始增大[26]. 即湖底和河流沉积物样品中的Ra同位素通过其母体(Th)的自然衰变而生成,通过解吸作用进入到间隙水中,然后在密度差的作用下向上覆水体扩散[29],在水体中垂直分布的表现为,Ra活度表层高,随深度增大而逐渐降低,达到一定深度,随深度的增大而增高[51]. 由于其半衰期很长,致使上覆水体中226Ra的添加十分缓慢,所以短时间内不会出现扩散输入与衰变之间的平衡,在图上的反映就是散点基本上呈现缓慢增加的趋势.
我们假设上覆水体中只存在沉积物扩散和放射衰变,并且沉积物释放出的Ra大于放射性衰变损失的量,也就是说在沉积物的上覆水体中Ra活度变化的结果是一个正值,那么这个值可以表示为:
dI/dt=Jdiff·Asurf-λ·I
(6)
通过积分可以得到:
I=(Jdiff·A-e-λ·t)/λ
(7)
而上覆水体中Ra同位素的活度随时间的变化可以表达为[52]:
It=(Jdiff·Adiff/λ)(1-e-λ·t)+I0·e-λ·t
(8)
式中,I0和It是初始时刻和t时刻上覆水体中Ra的库存量(dpm),Jdiff是单位面积、单位时间内Ra的扩散通量(dpm/(m2·h)),λ是镭的衰变常数(h-1),Adiff是培养样品的面积(河流=0.1m2,湖底沉积物=0.05m2). 对于长半衰期226Ra和228Ra来说,由于其半衰期很长,在培养时间范围的由放射衰变损失的量可以忽略. 因此226Ra和228Ra的活度随时间的变化公式为:
It=Jdiff·Asurf·t
(9)
根据理论公式(8)和(9)用拟合法来确定布哈河沉积物和青海湖底部沉积物培养样的Jdiff值,得到布哈河和青海湖湖底沉积物样品的Ra扩散速率(表2). 河流沉积物226Ra和228Ra扩散速率分别是0.039 和0.29dpm/(m2·h);湖底沉积物226Ra和228Ra扩散速率分别是0.018和0.092dpm/(m2·h). 河流底部沉积物中Ra的扩散速率要比湖底沉积物中Ra的扩散速率快,可能是因为湖泊底部表层沉积物长时间在咸水中,Ra同位素早已经发生解吸,沉积物中226Ra的扩散速率比228Ra小,是由于226Ra的再生速率很慢,上覆水体中226Ra的添加十分缓慢造成的.
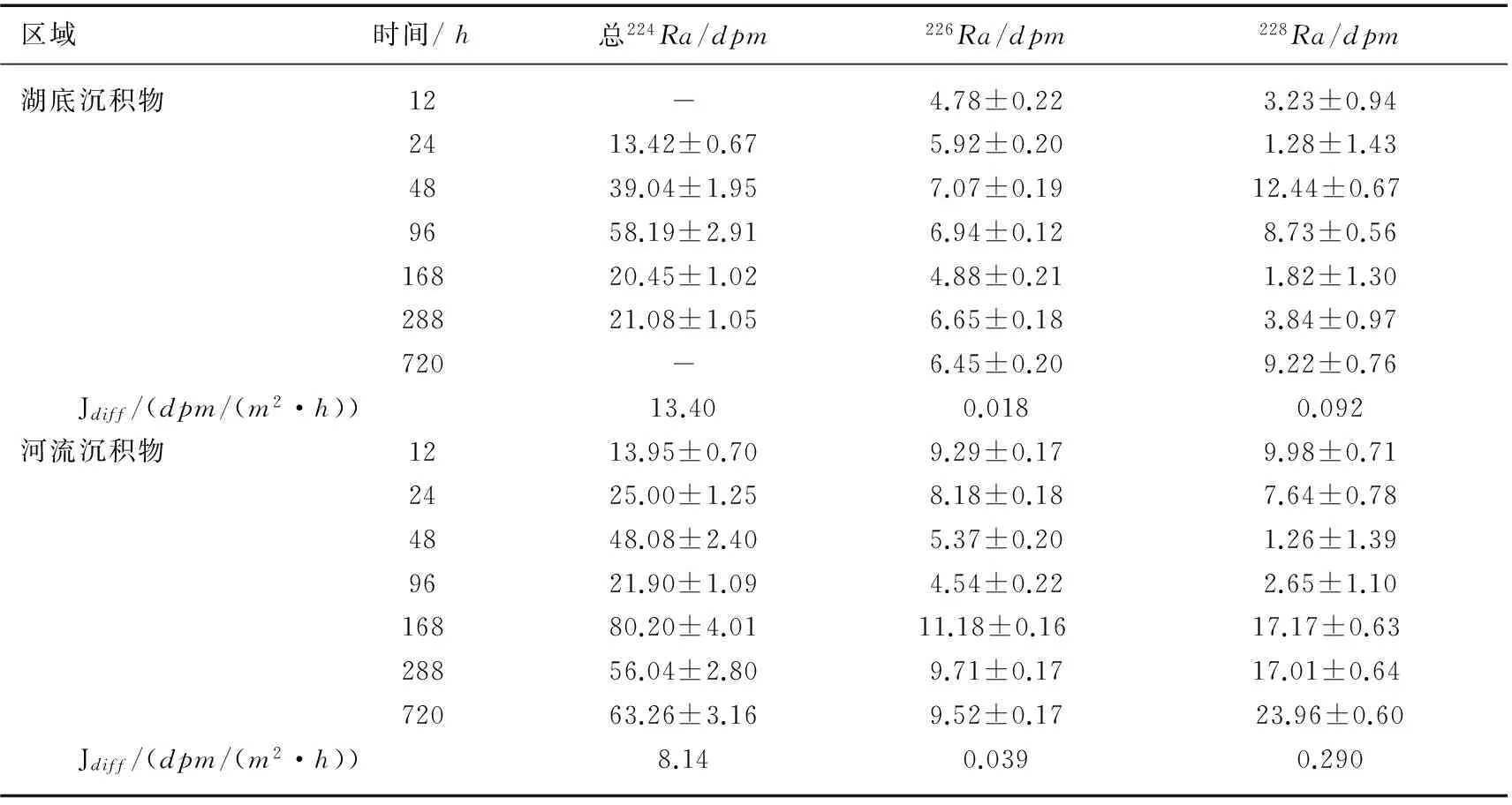
表2 布哈河河口和青海湖湖底沉积物中Ra同位素扩散通量Tab.2 Diffusion flux of Radium from sediments of Buha River estuary and Lake Qinghai
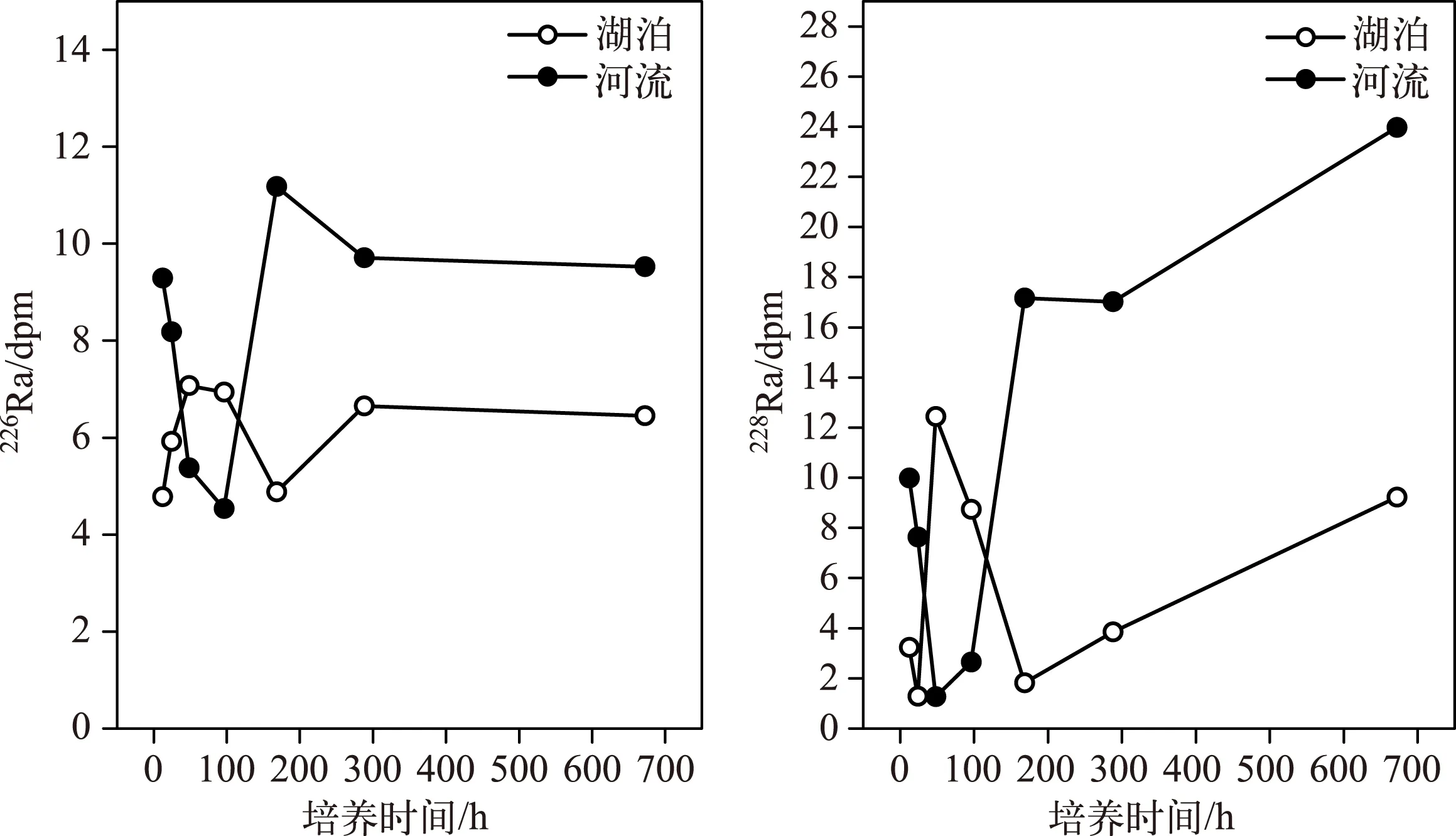
图5 布哈河河口沉积物中Ra扩散量与培养时间的关系Fig.5 Relationship between Radium diffusion flux in sediments of Buha River estuary and time
Garcia-Solsona等用意大利Venice瀉湖北部盐沼的沉积物做了扩散实验,得到沉积物中226Ra和228Ra的扩散通量分别为0.28和0.45Bq/(m2·d)[51]. 郭占荣等对九龙江河口沉积物进行了扩散实验,得到九龙江河口沉积物中226Ra的扩散通量为0.04Bq/(m2·d)[52].Beck等对Jamaica湾潮间带的沉积物镭扩散实验结果显示沉积物中223Ra和224Ra的扩散通量分别为0.02和0.47Bq/(m2·d),Jamaica湾水体中的Ra有4%~11%来源于海底沉积物的扩散输入[40]. 布哈河河口和青海湖湖底沉积物的226Ra和228Ra扩散速率与沿海地区相比较小,比胶州湾的略小,但是在同一个数量级上(表3).
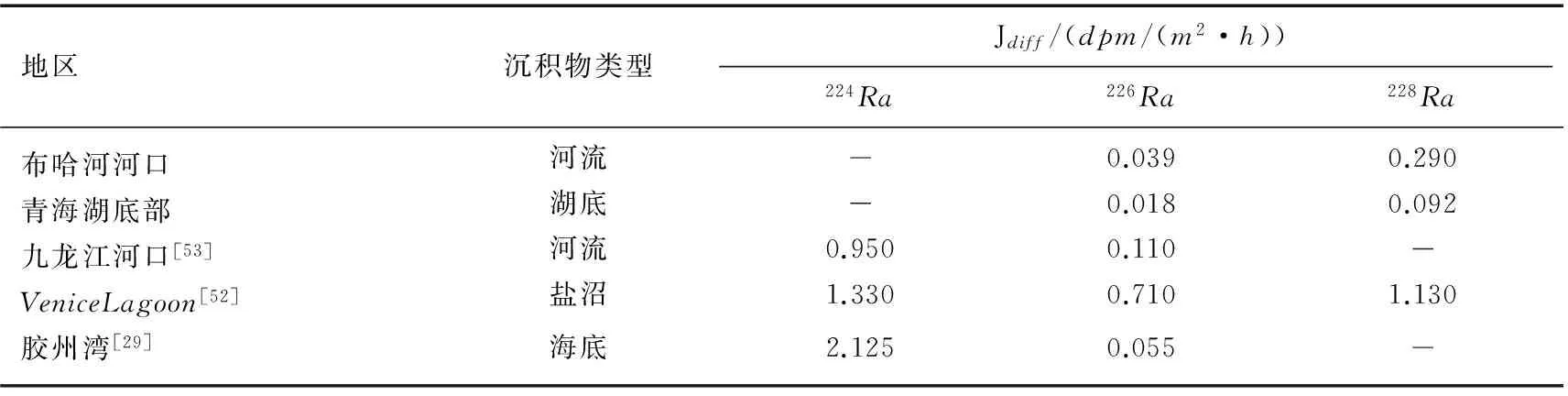
表3 青海湖沉积物中Ra的扩散速率与其他地区的对比Tab.3 Diffusion rate of Radium in sediments of Lake Qinghai compared with that of other areas
沉积物扩散出的镭同位素通量可以用下式进行计算:
Idiff=Jdiff·Ased·Hday
(10)
式中,Jdiff是单位面积、单位时间内沉积物扩散的镭通量(dpm/(m2·h)),Ased是表层沉积物的覆盖面积(m2),Hday是每天的小时数(24h).
研究区域青海湖南湾和北湾的水体覆盖面积分别为4.30×108和1.51×108m2. 根据公式(10)可以算出青海湖北湾湖底沉积物每天释放出的226Ra和228Ra通量分别为1.86×108和9.49×108dpm,南湾分别为6.52×107和3.33×108dpm,如果换成浓度的话,北湾和南湾通过湖底沉积物扩散的226Ra和228Ra的活度分别是0.005和0.025dpm/100L,布哈河集流面积为1.43×1010m2,同样根据公式(3)~(10)可以算出布哈河河底沉积物每天释放出的226Ra和228Ra通量分别为1.34×1010和9.95×1010dpm,换成浓度的话,通过河底沉积物扩散226Ra和228Ra的活度分别是1.7和12.6dpm/100L,分别占到河流溶解态226Ra和228Ra的8.0%和37.80%. 由此可见,青海湖湖底沉积物中Ra同位素扩散的活度与河流和地下水体中溶解态的Ra活度相比非常小,比误差值还小,所以湖底沉积物扩散的Ra活度几乎可以忽略,而布哈河河底沉积物扩散的Ra活度是不可以忽略的.
4 结论
1)通过对布哈河河口悬浮颗粒物中Ra进行解吸实验,得到了不同盐度(2.8‰、5.8‰、8.8‰、11.8‰和14.8‰)布哈河悬浮颗粒物中Ra的解吸活度, 其中223Ra、224Ra、226Ra和228Ra的解吸活度分别为0.018±0.001~0.069±0.003、0.32±0.02~0.59 ±0.03、0.13±0.017~0.29±0.014和0.11±0.02~0.23±0.03dpm/g.224Ra的解吸活度均高于226Ra和228Ra的解吸活度.
2)在固—液分配体系中随着盐度的逐渐增加,解吸态Ra同位素活度呈增加趋势,在盐度为12‰附近时布哈河悬浮颗粒物中226Ra和228Ra的解吸活度达到了最大值,当盐度<9‰时,226Ra的解吸活度大于228Ra,当盐度>9‰时,228Ra的解吸活度大于226Ra,这可能与当地富铀矿有关. 当盐度<4‰和盐度>12‰时,Ra的解吸量增加较为缓慢,当4‰<盐度<12‰时,Ra的解吸量增加较迅速.pH值的变化对青海湖Ra解吸的影响较大,并且比盐度对Ra解吸的影响更明显.
3)通过扩散实验,得到了布哈河和青海湖湖底沉积物中226Ra和228Ra的扩散速率. 河流沉积物中226Ra和228Ra的扩散速率分别是0.039和0.290dpm/(m2·h);湖底沉积物226Ra和228Ra的扩散速率分别是0.018 和0.092dpm/(m2·h). 湖底沉积物扩散活度小于河流沉积物扩散活度,在分析青海湖水体中Ra同位素的来源时,湖底沉积物扩散量可以忽略,而河流底部沉积物中Ra同位素的扩散量是不可以忽略的.
[1]Rama,MooreWS.MechanismoftransportofU-Thseriesradioisotopesfromsolidsintogroundwater. Geochimica et Cosmochimica Acta, 1984, 48(2): 395-399.
[2]PorcelliD,SwarzenskiPW.ThebehaviorofU-andTh-seriesnuclidesingroundwater. Reviews in Mineralogy and Geochemistry, 2003, 52(1): 317-361.
[3]SuksiJ,RasilainenK,CasanovaJet al.U-seriesdisequilibriainagroundwaterflowrouteasanindicatorofuraniummigrationprocesses. Journal of Contaminant Hydrology, 2001, 47(2): 187-196.
[4]ZhangLei.RadiumisotopesinChangjiangEstuary/EastChinaSeaandtheirapplieationinanalysisofingamongmultiplewatermasses[Dissertation].Hangzhou:ZhejiangUniversity, 2007 (inChinesewithEnglishabstract).[张磊. 长江口、东海的镭同位素及其在水团混合分析中的应用[学位论文]. 杭州: 浙江大学, 2007.]
[5]DulaiovaH,BurnettWC.EvaluationoftheflushingratesofApalachicolaBay,Floridavianaturalgeochemicaltracers. Marine Chemistry, 2008, 109(3): 395-408.[6]MooreWS.Determiningcoastalmixingratesusingradiumisotopes. Continental Shelf Research, 2000, 20(15): 1993-2007.
[7]MooreWS.Fifteenyearsexperienceinmeasuring224Raand223Rabydelayed-coincidencecounting. Marine Chemistry, 2008, 109(3): 188-197.
[8]PetersonRN,BurnettWC,TaniguchiMet al.DeterminationoftransportratesintheYellowRiver-BohaiSeamixingzonevianaturalgeochemicaltracers. Continental Shelf Research, 2008, 28(19): 2700-2707.
[9]BurnettW,AggarwalP,AureliAet al.Quantifyingsubmarinegroundwaterdischargeinthecoastalzoneviamultiplemethods. Science of the Total Environment, 2006, 367(2): 498-543.
[10]BurnettWC,PetersonR,MooreWSet al.Radonandradiumisotopesastracersofsubmarinegroundwaterdischarge-resultsfromtheUbatuba,BrazilSGDassessmentintercomparison. Estuarine, Coastal and Shelf Science, 2008, 76(3): 501-511.
[11]MooreWS.Largegroundwaterinputstocoastalwatersrevealedby226Raenrichments. Nature, 1996, 380(6575): 612-614.
[12]Rama,MooreWS.Usingtheradiumquartetforevaluatinggroundwaterinputandwaterexchangeinsaltmarshes. Geochimica et Cosmochimica Acta, 1996, 60(23): 4645-4652.
[13]MooreWS.Sourcesandfluxesofsubmarinegroundwaterdischargedelineatedbyradiumisotopes. Biogeochemistry, 2003, 66(1/2): 75-93.
[14]SwarzenskiPW,ReichC,KroegerKDet al.RaandRnisotopesasnaturaltracersofsubmarinegroundwaterdischargeinTampaBay,Florida. Marine Chemistry, 2007, 104(1/2): 69-84.
[15]SuN,DuJ,LiYet al.EvaluationofsurfacewatermixingandassociatednutrientfluxesintheEastChinaSeausing226Raand228Ra. Marine Chemistry, 2013, 156: 1-12.
[16]SuN,DuJZ,MooreWSet al.AnexaminationofgroundwaterdischargeandtheassociatednutrientfluxesintotheestuariesofeasternHainanIsland,Chinausing226Ra. Science of the Total Environment, 2011, 409(19): 3909-3918.
[17]KrestJM,HarveyJW.Usingnaturaldistributionsofshort-livedradiumisotopestoquantifygroundwaterdischargeandrecharge. Limnology and Oceanography, 2003, 48(1): 290-298.
[18]WebsterIT,HancockGJ,MurrayAS.Modellingtheeffectofsalinityonradiumdesorptionfromsediments. Geochimica et Cosmochimica Acta, 1995, 59(12): 2469-2476.
[19]DukatDA,KuehlSA.Non-steady-state210Pbfluxandtheuseof228Ra,226RaasageochronometerontheAmazoncontinentalshelf. Marine Geology, 1995, 125(3): 329-350.
[20]SuCC,HuhCA.210Pb,137Csand239,240PuinEastChinaSeasediments:sources,pathwaysandbudgetsofsedimentsandradionuclides. Marine Geology, 2002, 183(1): 163-178.
[21]WattersDL,KlineDE,CoaleKHet al.Radiometricageconfirmationandgrowthofadeep-watermarinefishspecies:Thebankrockfish,Sebastesrufus. Fisheries Research, 2006, 81(2): 251-257.
[22]KrestJM,MooreWS.226Raand228RainthemixingzonesoftheMississippiandAtchafalayaRivers:indicatorsofgroundwaterinput. Marine Chemistry, 1999, 64(3): 129-152.
[23]LiYH,MathieuG,BiscayePet al.Thefluxof226Rafromestuarineandcontinentalshelfsediments. Earth and Planetary Science Letters, 1977, 37(2): 237-241.
[24]LangmuirD,RieseAC.Thethermodynamicpropertiesofradium. Geochimica et Cosmochimica Acta, 1985, 49(7): 1593-1601.
[25]CableJE,SmithCG,BlanfordWJ.MeasurementsofdispersivityandretardationfactorsinmarinesedimentsusingtritiatedcalciumchloridesolutionandRadium-226. Radioprotection, 2009. 44(5): 185-190.
[26]GonneeaME,MorrisPJ,DulaiovaHet al.Newperspectivesonradiumbehaviorwithinasubterraneanestuary. Marine Chemistry, 2008, 109(3): 250-267.
[27]BeckAJ,CochranMA.Controlsonsolid-solutionpartitioningofradiuminsaturatedmarinesands. Marine Chemistry, 2013, 156: 38-48.
[28]SmithB,AmonetteA.Theenvironmentaltransportofradiumandplutonium:areview.Maryland:InstituteforEnergyandEnvironmentalResearch, 2006.
[29]YuanXiaojie,GuoZhanrong,LiuJieet al.Characteristicsofradiumdesorptionfromsedimentsinthesaltwaterenvironment. Acta Geoscientica Sinica, 2014, 35(5): 582-588 (inChinesewithEnglishabstract).[袁晓婕, 郭占荣, 刘洁等. 咸水环境下沉积物中镭的解吸特点. 地球学报, 2014, 35(5): 582-588.]
[30]WangR,ChauA,LiuFet al.Studiesontheadsorptionandmigrationofradiuminnaturalminerals. Journal of Radioanalytical and Nuclear Chemistry, 1993, 171(2): 347-364.
[31]SunY,TorgersenT.Adsorption-desorptionreactionsandbioturbationtransportof224Rainmarinesediments:aone-dimensionalmodelwithapplications. Marine chemistry, 2001, 74(4): 227-243.
[32]TurekianKK.Radiumonsoilmineralsurfaces:Itsmobilityunderenvironmentalconditionsanditsroleinradonemanation.Finalreport.YaleUniv.Fundingorganisation:USDOEOfficeofEnergyResearch,Washington,DC(UnitedStates):NewHaven,CT(UnitedStates), 1997.
[33]KrupkaK,SerneR.Understandingvariationinpartitioncoefficient,Kd,values,VolumeIII:ReviewofgeochemistryandavailableKdvaluesforamericium,arsenic,curium,iodine,neptunium,radium,andtechnetium.Richland:PacificNorthwestNationalLaboratory, 2000, 285: 271-277.
[34]VinsonDS.VengoshA,HirschfeldDet al.Relationshipsbetweenradiumandradonoccurrenceandhydrochemistryinfreshgroundwaterfromfracturedcrystallinerocks,NorthCarolina(USA). Chemical Geology, 2009, 260(3): 159-171.
[35]TomitaJ,ZhangJ,YamamotoM.Radiumisotopes(226Raand228Ra)inNa-CltypegroundwatersfromTohokuDistrict(Aomori,AkitaandYamagataPrefectures)inJapan. Journal Of Environmental Radioactivity, 2014, 137: 204-212.
[36]XuBochao.Preconcentrationanddeterminationofradiumisotopesandtheirapplicationsastracerstoassesswatermixingprocessesinestuaries[Dissertation].Qingdao:OceanUniversityofChina, 2011(inChinesewithEnglishabstract).[许博超, 天然镭同位素富集和测定方法及对河口混合过程的示踪研究[学位论文]. 青岛: 中国海洋大学, 2011.]
[37]MartinP,AkberRA.Radiumisotopesasindicatorsofadsorption-desorptioninteractionsandbariteformationingroundwater. Journal of Environmental Radioactivity, 1999, 46(3): 271-286.
[38]TsezosM,KellerD.Adsorptionofradium226bybiologicaloriginabsorbents. Biotechnology and Bioengineering, 1983, 25(1): 201-215.
[39]HuangLei.ResearehongroundwaterdischargeintoJiulongjiangEstuary[Dissertation].Xiamen:XiamenUniversity, 2009 (inChinesewithEnglishabstract).[黄磊. 九龙江河口区的地下水输入研究[学位论文]. 厦门: 厦门大学, 2009.]
[40]BeckAJ,TsukamotoY,Tovar-SanchezAet al.Importanceofgeochemicaltransformationsindeterminingsubmarinegroundwaterdischarge-derivedtracemetalandnutrientfluxes. Applied Geochemistry, 2007, 22(2): 477-490.
[41]LanzhouBranchChineseAcdemyofScienceseieneeed.EvolutionofrecentenvironmentinQinghailakeanditsprediction.Beijing:SciencePress, 1994: 270(inChinese).[中国科学院兰州分院. 青海湖近代环境的演化和预测. 北京: 科学出版社, 1994: 270.]
[42]SuNi.Tracingcoastalwatermixingprocessesandsubmarinegroundwaterdischargebyradiumisotopes[Dissertation].Shanghai:EastChinaNormalUniversity, 2013(inChinesewithEnglishabstract).[苏妮. 镭同位素示踪的近岸水体混合和海底地下水排泄[学位论文]. 上海: 华东师范大学, 2013.]
[43]ElsingerRJ,MooreWS.226Raand228RainthemixingzonesofthePeeDeeRiver-Winyahbay,YangtzeRiverandDelawarebayestuaries. Estuarine, Coastal and Shelf Science, 1984, 18(6): 601-613.
[44]ShiWenyuan,QiuXiaohui,HuangYipu.Thedistributionofsoluble226RainJiulongjiang-XiamenBay. Acta Oceanologica Sinica, 1993, 15(4): 50-55(inChinesewithEnglishabstract).[施文远, 邱晓晖, 黄奕普. 九龙江-厦门湾河口区溶解226Ra的分布. 海洋学报, 1993, 15(4): 50-55.]
[45]KiroY,YechieliY,VossCIet al.Modelingradiumdistributionincoastalaquifersduringsealevelchanges:TheDeadSeacase. Geochimica et Cosmochimica Acta, 2012, 88: 237-254.
[46]MooreWS.RadiumisotopesintheChesapeakeBay. Estuarine, Coastal and Shelf Science, 1981, 12(6): 713-723.
[47]KeyR,StallardR,MooreWet al.Distributionandfluxof226Raand228RaintheAmazonRiverestuary. Journal of Geophysical Research, 1985, 90(4): 6995-7004.
[48]AstwoodHM.ThedesorptionofradiumfromAmazonsediment[Dissertation].Columbia:UniversityofSouthCarolina, 1991.
[49]KongFC.Analysisofthedistributioncharacteristicsof226Raand228RaandtheirsourcesinthewesternpartofQinghaiLake. Chinese Journal of Oceanology and Limnology, 2015, 33(6): 1402-1412.
[50]MooreWS,SarmientoJL,KeyRM.Submarinegroundwaterdischargerevealedby228RadistributionintheupperAtlanticOcean. Nature Geoscience, 2008, 1(5): 309-311.
[51]Garcia-SolsonaE,MasquéP,Garcia-OrellanaJet al.EstimatingsubmarinegroundwaterdischargearoundIsolaLaCura,northernVeniceLagoon(Italy),byusingtheradiumquartet. Marine Chemistry, 2008, 109(3): 292-306.
[52]GuoZhanrong,HuangLei,YaungXiaojieet al.EstimatingsubmarinegroundwaterdischargetotheJiulongRiverestuaryusingRaisotopes. Advances in Water Science, 2011, 22(1): 118-125. (inChinesewithEnglishabstract).[郭占荣, 黄磊, 袁晓婕等. 用镭同位素评价九龙江河口区的地下水输入. 水科学进展, 2011, 22(1): 118-125.]
Desorption and diffusion characteristics of radium isotopes from particles in the western part of Lake Qinghai
KONG Fancui1, SHA Zhanjiang1,2**, DU Jinzhou3, SU Weigang4, HU Jufang1,5, WANG Qiugui1,5, MA Yujun2, ZHAI Yule2, WANG Zhuan2& MA Haiying6
(1: Qinghai Institute of Salt Lakes, Chinese Academy of Sciences, Xining 810008, P.R.China) (2: Key Laboratory of Natural Geography and Environmental Process in Qinghai Province Education, Qinghai Normal University, Xining 810008, P.R.China)
TwolaboratoryexperimentsforRadiumdesorptionanddiffusionwereconductedinthiswork,whichincluded1)theRadiumdesorptionofsedimentsthatwereunderdifferentsalinities(2.8‰, 5.8‰, 8.8‰, 11.8‰and14.8‰)fromthelakewaterandthepotentialofhydrogenfromBuhaRiverestuary. 2)Radiumdiffusivefluxeswithtimeintheoverlyingwateroflakesedimentsandinriversediments.WehavegotthedesorbedactivityofRadiumfromthesuspendedparticlesandRadiumdiffusionfluxesfromsurfacesediments.Theresultsshowthatthedesorptionactivitiesof224Raarehigherthanthoseof226Raand228Ra,andthedesorptiondegreesof224Ra,226Raand228Rahavereachedthemaximumvaluewhenthesalinitynear12‰.Thedesorptionof226Raactivitiesaregreaterthanthatof228Rawhensalinity<9‰,however,whensalinity>9‰,thedesorptionactivitiesof226Raislessthanthatof228Ra,whichmaybeassociatedwiththelocalrichuranium.Thediffusionfluxesof226Raand228RaofsedimentsfromBuhaRiverare0.039and0.290dpm/(m2·h),respectively,andtheyare0.018and0.092dpm/(m2·h)fromLakeQinghai,respectively.TheRadiumdiffusionfluxesofsedimentsinLakeQinghaiarelessthanthatoftheriver.
Radiumisotopes;desorption;diffusion;BuhaRiverestuary;LakeQinghai
*中国科学院百人计划项目“高原内陆盐湖水环境生物地球化学过程”、中国科学院青海盐湖研究所青年引导基金B类项目和青海省科技厅自然基金项目“大柴旦周围水体中锂的地球化学分布规律及对盐湖补给通量研究”(2016-ZJ-921Q)联合资助. 2015-08-21收稿; 2015-11-16收修改稿. 孔凡翠(1984~),女,博士,研究实习员;E-mail:kfc@isl.ac.cn.
**通信作者;E-mail: zhanjiang@sina.com.
