分散剂对柠檬酸络合法制备氧化镧钇亚微米粉体的影响
2016-10-12赵森龙张晓婷王焕平杨清华雷若姗徐时清
赵森龙,张晓婷,王焕平,杨清华,雷若姗,徐时清
(中国计量学院材料科学与工程学院, 杭州 310018)
分散剂对柠檬酸络合法制备氧化镧钇亚微米粉体的影响
赵森龙,张晓婷,王焕平,杨清华,雷若姗,徐时清
(中国计量学院材料科学与工程学院, 杭州310018)
以硝酸镧、硝酸钇和柠檬酸为原料,采用柠檬酸络合法制备氧化镧钇亚微米粉体,分别探讨了PEG400和油酸作为分散剂时对粉体物相、微观形貌以及粒径分布的影响规律。结果发现,无论是以PEG400还是油酸作为分散剂,煅烧后获得的粉体中只存在Y2O3单相,且粉体粒径随着煅烧温度的升高而变大;PEG400的最佳添加量为4wt%,此时粉体在1100 ℃煅烧后的一次粒径D50在85nm左右;油酸的最佳添加量为2wt%,此时粉体在1100 ℃煅烧后的一次粒径D50在75nm左右;使用油酸作为分散剂明显比使用PEG400作为分散剂的团聚峰平缓,说明油酸的分散效果比PEG400的分散效果更为优异。
氧化镧钇;PEG400; 油酸; 分散性
1 引 言
氧化镧钇透明陶瓷具有荧光性质、无双折射现象、低声子能量(约为370cm-1)、高熔点(大于2400 ℃)、高热导率和高顺磁性等特点,是一种性能优良的高温红外材料和电子材料;在添加Nd3+、Eu3+等稀土元素后,氧化镧钇透明陶瓷能产生高质量大功率的光束,是一种理想的固体激光器工作物质[1-6]。自从1981年Rhodes[7]在2150 ℃烧结、1900 ℃退火制备含La2O3的Y2O3透明陶瓷以来,氧化镧钇透明陶瓷的光学性能、热力学性能,以及烧结工艺与各种性能的内在联系规律已成为研究热点。近年来,以上海大学杨秋红教授为首的课题组,在国际上首次实现了掺镱氧化镧钇透明陶瓷的激光输出[8]。
影响陶瓷透明度的因素有很多,其中粉体的晶体结构与缺陷、粉体的纯度和粒度、粉体的分散性等因素都会影响到陶瓷的透明度。因此,粉体的制备是生产高质量透明陶瓷的前提和关键,颗粒大小均匀、形状一致、分散性好的单相亚微米甚至纳米级粉体对透明陶瓷的制备具有重要意义[9]。柠檬酸盐法具有操作简单、设备要求低、生产成本低、适合多组分体系、可达到纳米级别、预烧过程不产生中间相等优点,是制作透明激光陶瓷粉体的常用方法[10,11]。但是,细小粉体颗粒的表面活性使其很容易团聚在一起从而形成带有若干连接界面的尺寸较大的团聚体,致使粉体出现流动性差、烧结后存在大量气孔等缺点[12]。为了减轻亚微米粉体间的团聚现象,在粉体制备过程中引入分散剂是最为有效的方法,如聚乙二醇、油酸、甲基丙烯酸和丙烯酸等已经用于分散不同类型的粉体[13-15];由于不同分散剂作用机理的差异,选择合适的分散剂种类及用量对减轻粉体的团聚尤为重要。本文分别选取PEG400和油酸作为分散剂,通过改变其添加量来确定合适的分散剂种类及用量,制备出分散性良好的氧化镧钇亚微米粉体,为高质量透明陶瓷的制备奠定基础。
2 实 验
2.1样品制备
以高纯Y(NO3)3·6H2O(99.99%)、La(NO3)3·6H2O(99.99%)为原料,按化学式(Y0.9La0.1)2O3称量配制;同时加入无水柠檬酸(分析纯)和去离子水,其中金属阳离子摩尔数:柠檬酸摩尔数:去离子水摩尔数为1∶1.5∶15;分别加入PEG400和油酸作为分散剂。PEG400的添加量分别为溶液质量的0.5wt%、1wt%、2wt%、4wt%、8wt%、16wt%;油酸的添加量分别为溶液质量的1wt%、2wt%、4wt%、8wt%。将配好的溶液进行加热搅拌,直至溶液变得非常粘稠。将得到的粘稠溶胶在140 ℃干燥36h,得到疏松多孔的干凝胶;用研钵磨细后在井式炉中进行煅烧,其温度设置为1 ℃/min缓慢上升,在600 ℃保温4h排除掉有机物质,然后继续升温至1000 ℃、1100 ℃或1200 ℃保温2h进行煅烧。
2.2样品测试
用德国Burker公司BrukerAxsD2X射线衍射仪(XRD)分析粉体的物相组成。用日本HITACHI公司SU8010冷场发射扫描电子显微镜(SEM)观察粉体颗粒形貌及尺寸。用上海必能信超声有限公司的SB2200超声波发生器对粉体进行分散,然后用美国Microtrac公司S3500激光粒度分析仪测定样品的粒度分布。
3 结果与讨论
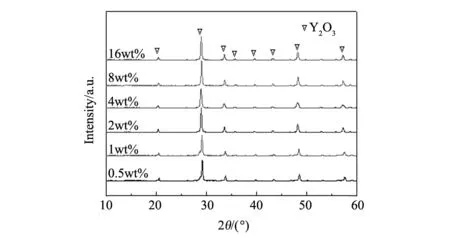
图1 不同PEG400添加量时氧化镧钇在1100 ℃煅烧后的XRD图谱Fig.1 XRD patterns of (Y0.9La0.1)2O3 powders calcined at 1100 ℃ with different content of PEG400
3.1PEG400的影响
图1示出了不同PEG400添加量时氧化镧钇在1100 ℃煅烧后的XRD图谱。从图中可以看出,1100 ℃煅烧后的氧化镧钇粉体只存在Y2O3单相,说明此时La2O3已经完全固溶在Y2O3里面。从图中还可以发现,随着PEG400添加量的增加,衍射峰的位置和强度几乎没有变化,这说明PEG400添加量的变化对氧化镧钇粉体的物相结构没有影响。
图2和图3分别示出了不同PEG400添加量时氧化镧钇在1100 ℃煅烧后的扫描电镜照片和激光粒度分析曲线。从图2中可以观察到,随着PEG400添加量的增加,粉体颗粒有先变小后不变的趋势。其中PEG400添加量为0.5wt%和1wt%时,粉体颗粒较大,在50~120nm之间。当PEG400的添加量增加到2wt%时,粉体颗粒有比较明显的变小,其尺寸在40~100nm之间。当PEG400的添加量继续增加时,粉体的颗粒尺寸变化不大。

图2 不同PEG400添加量时氧化镧钇在1100℃煅烧后的扫描电镜照片Fig.2 SEM images of (Y0.9La0.1)2O3 powders calcined at 1100 ℃ with the mass percent of PEG400 of (a)0.5wt%;(b)1wt%;(c)2wt%;(d)4wt%;(e)8wt%;(f)16wt%
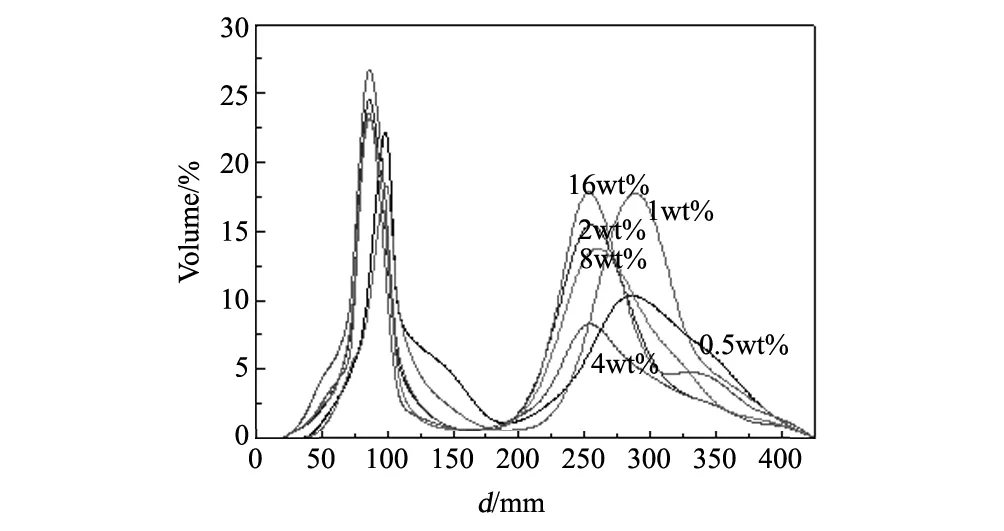
图3 不同PEG400添加量的氧化镧钇粉体在1100 ℃烧结后的激光粒度曲线Fig.3 Size distribution curve of (Y0.9La0.1)2O3powders calcined at 1100 ℃ with different content of PEG400

图4 PEG400添加量为4 wt%时氧化镧钇在不同温度煅烧后的XRD图谱Fig.4 XRD patterns of (Y0.9La0.1)2O3 powders of 4wt% PEG400 calcined at different temperatures
从图3中可以看出,随着PEG400添加量的增加,粉体的中心粒径尺寸有先变小后不变的趋势。其中PEG400的含量为0.5wt%和1wt%时,粉体的颗粒尺寸较大,其中心粒径(D50)在100nm左右,并且它们在300nm左右有明显的团聚现象;当PEG400含量为0.5wt%时,其团聚峰高度约为一次粒径主峰高度的1/2左右。当PEG400的添加量增加到2wt%和4wt%时,其颗粒尺寸有所变小,一次粒径的D50在85nm左右,并且其团聚现象有明显的减轻,如PEG400添加量为4wt%的团聚峰高度减小到了一次粒径主峰高度的1/4。当PEG400的添加量继续增加时,其一次粒径的中心粒径(D50)几乎没有变化,均在85nm附近,但其团聚现象又变得严重,如PEG400添加量为16wt%时,其团聚峰高度上升到一次粒径主峰高度的2/3。
聚乙二醇是一种非离子型分散剂,其分子式H-(O-CH2-CH2)n-OH中含有羟基和醚键两种亲水基,所以有很好的水溶性,易通过氢键使高分子长链的一端紧密地吸附在溶液前驱体表面,另一端则在溶液中伸展,形成一层高分子吸附层[16]。当2个带有高分子吸附层的质点相互靠近时,彼此间的吸附只是受挤压而不能穿透,由于空间限制使高分子链采取的可能构型数减少、构型熵减少,会使体系的自由能增加而产生排斥作用,从而使质点稳定,称为“空间位阻作用”[17,18]。当PEG400添加量为4wt%时,由于溶液前驱体表面形成了完整的高分子吸附层,从而有效地阻止了粉体的团聚,粉体颗粒较小在40~100nm之间。而当PEG400含较少时,如图2a的PEG400添加量为0.5%时,由于不能形成完整的高分子吸附层,从而使同一高分链两端吸附不同的溶液前驱体,使溶液前驱体之间相互靠近(“架桥作用”)[19],不能达到分散的目的,所以其粉体颗粒较大,在50~120nm左右。但是当加入大量的PEG400时,如图3中的PEG400添加量为16wt%时,其团聚现象明显增加,这是由于伸展在溶液中的高分子链一端相互缠绕在一起,增加了粉体的团聚。
图4和图5分别示出了PEG400添加量为4wt%时,氧化镧钇在不同温度煅烧后的XRD图谱和扫描电镜照片。从图4中可以看出,PEG400添加量为4wt%时,氧化镧钇分别在1000 ℃,1100 ℃和1200 ℃煅烧后的粉体都形成了Y2O3单相,说明此时La2O3已完全固溶入Y2O3里面。从图4中还可以发现,随着温度的升高,衍射峰的强度逐渐增强,且其衍射峰的位置不变,说明随着温度的升高,晶型并未发生变化且其晶体结构逐渐趋于完整。从图5中可以看出,PEG400添加量为4wt%时,粉体颗粒随着温度的升高而变大;在1000 ℃煅烧后的粉体颗粒尺寸在20~70nm之间,在1100 ℃煅烧后的粉体颗粒尺寸在40~100nm之间,在1200 ℃煅烧后的粉体颗粒尺寸在50~150nm之间。

图5 PEG400添加量为4wt%时氧化镧钇在不同温度煅烧后的扫描电镜照片Fig.5 SEM images of (Y0.9La0.1)2O3 powders of 4wt% PEG400 calcined at (a)1000 ℃;(b)1100 ℃;(c)1200 ℃
图6示出了PEG400添加量为4wt%时氧化镧钇在不同温度煅烧后的激光粒度分析曲线。从图中可以看出,随着煅烧温度的升高,粉体颗粒逐渐变大。当煅烧温度为1000 ℃时,其一次粒径的中心粒径(D50)在50nm左右,但是团聚较严重,其团聚峰在170nm左右,其高度约为一次粒径主峰高度的1/2。当煅烧温度上升到1100 ℃时,其颗粒尺寸有所变大,一次粒径的D50在85nm左右,而且其团聚现象也明显减轻,团聚峰高度降低至一次粒径主峰高度的1/4左右。当煅烧温度增加到1200 ℃时,其颗粒尺寸继续增大,一次粒径的D50在100nm左右,其结论与扫描电镜观察到的结果基本一致。
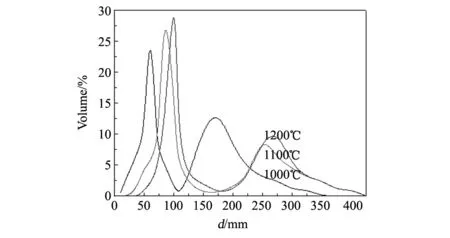
图6 PEG400添加量为4 wt%时氧化镧钇在不同温度煅烧后的激光粒度曲线Fig.6 Size distribution curve of (Y0.9La0.1)2O3powders 4wt% PEG400 by laser size detector calcined at different temperatures

图7 不同油酸添加量时氧化镧钇在1100 ℃煅烧后的XRD图谱Fig.7 XRD patterns of (Y0.9La0.1)2O3powders calcined at 1100 ℃ with different content of oleinic acid
3.2油酸的影响
图7示出了添加不同油酸含量时氧化镧钇在1100 ℃煅烧后的XRD图谱。从图中可以看出,1100 ℃煅烧后的氧化镧钇粉体只有Y2O3单相,说明此时La2O3已经完全固溶在Y2O3里面。从图中还可以发现,随着油酸含量的增加,衍射峰的位置和强度几乎没有变化,这说明油酸添加量的变化对氧化镧钇粉体的物相结构没有影响。
图8和图9分别示出了不同油酸含量时氧化镧钇在1100 ℃煅烧后的扫描电镜照片和激光粒度分析曲线。从图8中可以观察到,随着油酸添加量的增加,粉体颗粒有先变小后不变的趋势。其中当油酸添加量为1wt%时,粉体的颗粒比较大,在50~120nm之间。当油酸添加量增加到2wt%时,粉体颗粒有比较明显的变小,其尺寸在40~100nm之间。当油酸的含量继续增加时,粉体的颗粒尺寸变化不大。

图8 不同油酸添加量时氧化镧钇在1100℃煅烧后的扫描电镜照片Fig.8 SEM images of (Y0.9La0.1)2O3 powders calcined at 1100 ℃ with the mass percent of oleinic acid of (a)1wt%;(b)2wt%;(c)4wt%;(d)8wt%
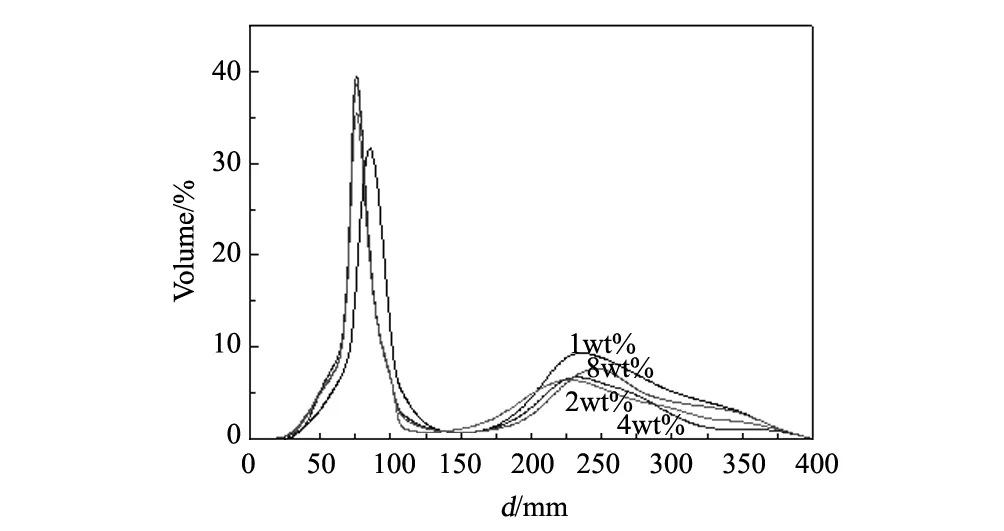
图9 不同油酸添加量的氧化镧钇粉体在1100 ℃烧结后的激光粒度曲线Fig.9 Size distribution curve of (Y0.9La0.1)2O3powders calcined at 1100 ℃ by laser size detector with different content of oleinic acid

图10 油酸添加量为2wt%时氧化镧钇在不同温度煅烧后的XRD图谱Fig.10 XRD patterns of (Y0.9La0.1)2O3 powders of 2wt% oleinic acid calcined at different temperatures
从图9中可以看出,随着油酸添加量的增加,粉体的中心粒径尺寸呈现先变小后不变的趋势。当油酸的添加量为1wt%时,粉体的颗粒尺寸较大,其一次粒径的中心粒径(D50)在90nm左右,并且在230nm左右有明显的团聚现象,其团聚峰高度占了一次粒径主峰高度的1/4左右。当油酸添加量增加到2wt%时,其颗粒尺寸明显变小,一次粒径的D50在75nm左右,并且团聚现象有明显的减轻,其团聚峰较平缓,高度仅占一次粒径主峰高度的1/8左右。当油酸的添加量继续增加时,其一次粒径的D50几乎没有变化,但团聚现象有所增加,如油酸添加量为8wt%时,其一次粒径的D50仍在75nm左右,但其团聚峰高度上升到一次粒径主峰高度的1/4。
油酸的分散作用机理与PEG400类似,其分子中的羧基通过化学吸附在前驱体表面形成吸附层,由于其“空间位阻作用”能很好地达到分散效果。当油酸含量较为合适时(2wt%),氧化镧钇粉体颗粒比较小,其尺寸在40~90nm之间,如图8b中所示;当油酸含量不够时,如图8a的油酸添加量为1wt%时,粉体的颗粒较大,尺寸在50~120nm之间,这主要是由于油酸不能完全包覆前驱体表面,使两个前驱体之间通过“架桥作用”连接,从而加重了团聚。同时,通过对图9和图3的比较,可以发现图9中的团聚峰明显比图3中的团聚峰低,说明使用油酸作为分散剂比使用PEG400的效果要好,这是因为油酸中含有的羧基比PEG400中含有的羟基活性更高,在溶液中可以与前驱体结合更紧密,不容易使高分子从前驱体表面脱落[20]。

图11 油酸添加量为2wt%时氧化镧钇在不同温度煅烧后的扫描电镜照片Fig.11 SEM images of (Y0.9La0.1)2O3 powders of 2wt% oleinic acid calcined at (a)1000 ℃;(b)1100 ℃;(c)1200 ℃

图12 油酸添加量为2wt%时氧化镧钇在不同温度煅烧后的激光粒度曲线Fig.12 Size distribution curve of (Y0.9La0.1)2O3powders 2wt% oleinic acid by laser size detector calcined at different temperatures
图10和图11分别示出了油酸添加量为2wt%时氧化镧钇在不同温度煅烧后的XRD图谱和扫描电镜照片。从图10中可以观察到,氧化镧钇分别在1000 ℃,1100 ℃和1200 ℃煅烧后的粉体都形成了Y2O3单相,说明此时La2O3已完全固溶在Y2O3里面。从图10中还可以发现,随着温度的升高,衍射峰的强度逐渐增强,且其衍射峰的位置不变,说明随着温度的升高,晶型并未发生变化且其晶体结构逐渐趋于完整。从图11中还可以观察到,粉体颗粒随着煅烧温度的升高而变大;在1000 ℃、1100 ℃、1200 ℃煅烧后的粉体颗粒尺寸分别在20~70nm、40~100nm、50~150nm之间。
图12示出了油酸添加量为2wt%时氧化镧钇在不同温度煅烧后的激光粒度分析曲线。从图中可以看出,随着煅烧温度的升高,粉体颗粒逐渐变大。当煅烧温度为1000 ℃时,其颗粒尺寸较小,一次粒径的中心粒径(D50)在60nm左右,但是团聚较为严重,其团聚峰在175nm左右、高度占了一次粒径主峰高度的1/3。当煅烧温度上升到1100 ℃时,其颗粒尺寸有所变大,一次粒径的D50在75nm左右,而且其团聚现象也明显减轻。当煅烧温度增加到1200 ℃时,其颗粒尺寸继续增大,一次粒径的D50在100nm左右,这与扫描电镜观察到的结果基本一致。
4 结 论
以硝酸镧、硝酸钇和柠檬酸为原料制备氧化镧钇亚微米粉体时,无论是以PEG400还是油酸作为分散剂,在1000 ℃、1100 ℃和1200 ℃煅烧后获得的粉体中只存在Y2O3单相,且粉体粒径随着煅烧温度的升高而变大;
以PEG400作为分散剂时,粉体的团聚现象随着PEG400添加量的增加,呈现先减轻后增加的趋势。PEG400的最佳添加量为4wt%,此时粉体在1100 ℃煅烧后的一次粒径D50在85nm左右,且在1000 ℃、1100 ℃和1200 ℃煅烧后的颗粒尺寸分别在20~70nm、40~100nm、50~150nm之间;
以油酸作为分散剂时,其效果要明显优于PEG400。油酸的最佳添加量为2wt%,此时粉体在1100 ℃煅烧后的一次粒径D50在75nm左右,且其团聚峰明显比使用PEG400作为分散剂时的团聚峰平缓。
[1] 李文杰,林辉,滕浩,等.Yb3+,Ho3+共掺氧化钇透明陶瓷的制备及其性能[J].无机化学学报,2010,26(4):687-692.
[2] 杨秋红,丁君,豆传国,等.用纳米粉制备Nd:Y1.84La0.16O3透明陶瓷[J].硅酸盐学报,2007,35(6):755-759.
[3] 卢歆.Yb3+:Y2O3透明陶瓷超细粉体的制备及性能研究[D].长春:长春理工大学工学硕士学位论文,2008.
[4]WangNL,ZhangXY,QiuGM,etal.SynthesisofLa3+andNd3+co-dopedyttriananopowderfortransparentceramicsbyoxalateprecipitationmethod[J].Journal of Rare Earths,2010,28(2):232-236.
[5]HuangYH,JiangDL,ZhangJX,etal.Synthesisofmono-dispresedsphericalNd:Y2O3poederfortransparentceramics[J].Ceramics International,2011,37:3523-3529.
[6] 孙晶,万玉春,姜涛,等.低温燃烧法制备Nd:Y2O3超细粉及性能研究[J].长春理工大学学报,2008,31(4):32-34.
[7]RhodesWH.Controlledtransientsolidsecond-phasesinteringofyttria[J].J. Am. Ceram Soc,1981,64(1):13-19.
[8] 杨秋红.我国掺钕氧化镧钇激光透明陶瓷实现激光输出[J].稀土信息,2009,(11):28.
[9] 王佳,吴起白,张海燕.Yb:YAG激光透明陶瓷纳米粉体的制备研究[J].陶瓷学报,2012,33(2): 127-132.
[10] 苏言杰,张德,徐建梅,等.柠檬酸盐凝胶自燃法合成超细粉体[J].材料导报, 2006, 20(4): 142-144.
[11] 陈松林,曾鲁举,曾大凡,等.燃烧合成理论基础和合成SiC/Si3N4复相原料进展[J].陶瓷学报, 2014, 35(2): 125-129.
[12] 李爱民,孙康宁,尹衍升,等.纳微米复合HAp-ZrO2生物复合材料的制备与微观结构研究[J].人工晶体学报,2003,32(6): 563-568.
[13] 赵娜如,康海峰,刘梦姣,等.pH值及分散剂对沉淀法制备β-磷酸三钙粉体性能的影响[J].硅酸盐通报, 2010, 29(1): 49-53.
[14]ChengLJ,KangY.SelectivepreparationofBi2O3visiblelight-drivenphotocatalystbydispersantandcalcinations[J].Journal of Alloys and Compounds, 2014, 585: 85-93.
[15] 谭训彦,尹衍升,李嘉,等.Fe-Al/ZrO2复合粉体的均匀分散[J].硅酸盐学报,2004,32(7): 827-831.
[16] 郝成伟,吴伯麟,李继彦.聚乙二醇分散剂对高纯细α-Al2O3制备的影响[J].材料导报, 2007,21(8): 163-167.
[17] 郑仕远,吴奇财,周朝霞,等.超细粉体的水性超分散剂研究进展[J].涂料工业,2011,41(5):74-79.
[18] 范金山.微乳液法制备TiO2纳米粉体及其光催化性能研究[J].人工晶体学报,2006,35(2):174-178.
[19] 徐晓娟,孙一宁,胡倩,等.聚乙二醇对单分散球形氧化铝前驱体的影响[J].辽宁大学学报, 2014,41(2):122-125.
[20] 李桂英,孔振兴,戴子林.氢氧化铝粉体表面改性的研究[J].材料研究与应用,2012,6(1):41-44.
InfluenceofDifferentDispersantontheDispersityof(Y0.9La0.1)2O3PowdersPreparedbyCitrateMethod
ZHAO Sen-long,ZHANG Xiao-ting,WANG Huan-ping,YANG Qing-hua,LEI Ruo-shan,XU Shi-qing
(CollegeofMaterialsScienceandEngineering,ChinaJiliangUniversity,Hangzhou310018,China)
The(Y0.9La0.1)2O3powderswerepreparedbythecitratemethodusingY(NO3)3·6H2O,La(NO3)3·6H2O,citricacidanddifferentdispersantastherawmaterials.Theinfluenceofdifferentdispersantandtemperaturewerediscussedonthephasestructure,sizeanddispersityofthepreparedpowders.Itisexhibitedthat,nomatterusingPEG400oroleinicacidasthedispersant,thepreparedpowdershaveasinglephaseofY2O3andthesizeofthepowdersbecomebigwiththecalcinationtemperaturesrise.ThebestmasspercentofPEG400is4wt%,andtheprimaryparticlesizeofthepowderscalcinedat1100 ℃isabout85nm,whilethebestmasspercentofoleinicacidis2wt%,andtheprimaryparticlesizeofthepowderscalcinedat1100 ℃isabout75nm.ItisalsoexhibitedthatthedispersityofthepowderswhichusingtheoleinicacidasthedispersantisbetterthanPEG400.
(Y0.9La0.1)2O3;PEG400;oleinicacid;dispersity
浙江省自然科学基金重点项目(LZ14B010001)
赵森龙(1991-),男,硕士研究生.主要从事纳米粉体及功能陶瓷方面的研究.
王焕平/徐时清,博士,教授.
O613
A
1001-1625(2016)01-0285-07
