面向疾病分类的人类互作网络拓扑模块的功能同质性分析
2016-09-29高盼盼王宁周雪忠刘光明王惠欣
高盼盼 王宁 周雪忠 刘光明 王惠欣


摘要:鉴于网络医学中尚未有对疾病分类与功能蛋白模块功能同质性分析之间关系的研究,展开以下研究工作:首先,利用Mesh、String9等数据库中的数据构建了基因关系网络;其次,采用基于优化模块度的模块划分方法(如BGLL、非负矩阵分解(NMF)等聚类算法)对基因关系网络进行了划分;再次,对划分出来的模块进行了GO富集分析,通过对高致病拓扑模块和低致病拓扑模块的GO富集分析的比较,发现了疾病分类和蛋白模块功能特性在生物过程、细胞组分、分子功能等方面存在重要的生物学提示;最后,分析了疾病分类的拓扑模块的功能特性,通过对网络拓扑性质如平均度、密度、平均最短路径长度等方面的分析得到了各模块的功能特点数据,进一步揭示了疾病分类和功能模块之间的相关关系。
关键词:网络医学;疾病分类;GO富集分析;蛋白功能模块;拓扑模块;Mesh;String9
中图分类号:TP399
文献标志码:A
0引言
近年来,随着复杂网络在生物医学领域的应用逐步广泛,网络医学为疾病分子层面的研究提供了平台。随着遗传学和基因组学的进步和发展,高通量蛋白质相互作用数据的产生推进了疾病和致病基因之间的关联研究。由于人类细胞内的分子组件间在功能上具有相互依赖性,所以很少有疾病是由单一基因异常而导致的结果,现在大量的研究发现疾病是各组织器官系统间的细胞内和细胞间各种复杂的网络的局部异常导致的。所以网络医学的发展不仅方便研究者可以借助生物分子网络比较系统地去探索导致某一疾病的分子复杂性,也可用于探索截然不同的表型疾病间潜在的分子关系,同时网络医学的研究发展也可以用于识别疾病目前尚未发现的致病基因。
网络医学在最近几十年已经得到越来越多的生物医学研究者的关注。Barabasi等[1]对人类疾病网络的特性进行了相关的研究,提出了拓扑模块、功能模块、疾病模块这三种模块存在共同的元素,同时还分析了如何利用网络结构进行基因预测,提出了基于边的方法、疾病模块的方法和随机传播算法;Sharan等[2]从蛋白质的近邻分析、马尔可夫随机场和基于模块的方法三个方面进行了蛋白质功能的预测;Lin等[3]对扩张心脏病进行了动态蛋白致病模块的分析,提出了一种新的框架来分析蛋白质相互作用(Protein-Protein Interaction,PPI)数据,并且成功找到了该病的蛋白功能模块。
我们发现上述研究主要集中在:对单个蛋白功能的预测,预测某个疾病的致病基因,为特定疾病寻找致病模块。目前还没有针对疾病分类进行功能蛋白模块的研究。通用的疾病分类体系是Mesh主题词分类,Zhou等[4]构建了人类疾病的表型网络并进行了详细的分析。在网络医学领域中还存在着如下的问题:
1)蛋白模块在基因本体(Gene Ontology,GO)上的分析;
2)疾病分类与蛋白功能模块关联关系的研究;
3)网络拓扑特性与疾病分类之间的关系。
本文将在人类蛋白相互作用网络基础上运用基于模块度的优化算法,如:BGLL[5]、非负矩阵分解(Nonnegtive Matrix Factorization, NMF),从Mesh疾病分类的角度结合拓扑模块富集的GO术语、致病基因相对比Ratio值以及一些常用的拓扑特性对人类疾病分类和人类互作网络拓扑模块的功能进行同质性分析。
1基本概念及常用聚类算法
2数据的来源及整理
采用Mesh给出的疾病名称为标准名称,基因部分采用National Center for Biotechnology Information(NCBI)提供的人类基因数据,Comparative Toxicogenomics Database(CTD)、ClinVar、DisGeNet和DiseaseConnect四个数据源整合疾病与基因之间的关系,最后去掉重复数据,得到137308条关系数据,包括2896个疾病及15735个基因。
STRING 9.1 提供了蛋白质与蛋白质相互作用关系,并使用Score值量化,筛选出与人类有关且Score大于700的记录,然后将其映射到NCBI中,最终得到436326条记录,基因数是13734。基因关系网络生成过程如图1。
3疾病分类相关拓扑模块的GO功能分析
本章利用BGLL和NMF算法对基因关系网络进行划分,通过BGLL划分后得到314个模块,通过NMF得到301个模块,并计算了两种方式划分下的模块的一致性,结果如图2所示。所谓模块的一致性就是指用不同的划分方法划分网络,然后计算模块的一致性,也就是说对于基因A、B、C,用BGLL划分在一个模块中,而用NMF也被划分也在一个模块中,这就叫模块的一致性。
从图2中可以看到,用不同方法划分的模块的一致性大于0.6的有78.095%,可以认为用不同聚类算法得到的模块具有一致性。下面只对BGLL划分的模块进行详细的疾病分类和蛋白质功能同质性分析。
3.1基于人类互作网络的BGLL社区划分
使用BGLL算法将基因基因网络进行社团划分,得到314个拓扑模块。该算法的最终划分结果的模块度是0.378,模块划分算法比较合理。图3是模块4的网络结构示意图。
分析图4、5可以发现,人类疾病在拓扑模块功能方面有明显的表达倾向,人类疾病与拓扑模块之间的多样性一致,通过社团划分方法得到的人类互作网络拓扑模块能够有效地反映出其在人类疾病方面的功能性特征。
3.3高致病拓扑模块的GO富集分析
为了更好地探究人类疾病与网络划分的拓扑模块之间的关系,从Ratio特殊值入手,分析拓扑模块在人类疾病表达中所起的作用。由3.2节可知,Ri=1.409的模块{55,59,82,95,102,109,111,123,127,132,144,163,192,201,218,232,237,250,251,257,261},表示模块Mi的基因全是致病基因,将Ri=1.409的模块称为“高致病拓扑模块”。然后对这些模块进行GO富集分析,表1是对这些模块的基因富集在GO术语上的情况,其中“null”代表这些模块没有富集到GO term上,故其后一列的P-value无计算值。
观察表1可知,模块95和模块257在GO上三个分支的富集P-value都大于0.01,即GO富集效果不好,因此说明这两个模块在GO上没有进行显著的富集。但是除此以外,其他的模块的GO富集效果都比较好,而且这些模块大部分都与重要的功能特性相关,并且在基因的表达和拓扑模块功能性的表达方面也有重要的表现。因此可以说明致病基因相对比值(Ratio)较大时,拓扑模块的功能与人类疾病紧密相关。
表2~4分别是从生物过程(biological process, bp)、细胞组分(cellular component, cc)、分子功能(molecular function, mf)三个方面对高致病拓扑模块的GO分析的部分模块在GO术语上的情况的结果展示。从表中可以看出,这些拓扑模块富集的GO与重要的功能特性相关,比如模块55,生物过程方面,该拓扑模块富集到GO:0006590,且P-value为3.91E-10,远小于0.01,很有代表性,表明模块55是与甲状腺激素产生的过程有密切联系的功能模块;在细胞组分方面,拓扑模块富集到GO:0016021,且P-value为0.005631863,小于0.01,与整合膜有密切关系;分子功能方面,拓扑模块富集到GO:0016174,P-value为3.09E-05,远小于0.01,在分子功能NAD(P)H氧化酶发挥作用中起到至关重要的作用。
如果模块55的基因发生突变,将对与之紧密相关的生物过程、细胞组分、分子功能产生影响,因而人类疾病与55模块有很大可能性的关联。其他高致病拓扑模块也有类似的关系,因此可以说明,拓扑模块与重要的生物功能特性有密切关联。
3.4低致病拓扑模块的GO富集分析
本节分析Ratio的取值等于0的模块在人类疾病表达中所起的作用。模块{155,158,260,262,280,286,296,297,
311,312,313}致病基因相对比值Ri=0,表示拓扑模块的基
因全部为非致病基因,称这些模块为低致病拓扑模块。然后对这些模块进行GO富集分析,以便去探究拓扑模块内基因富集在GO术语上的情况。GO分析从生物过程、细胞组分、分子功能三个方面进行,表5中P-value加下划线的值是小于0.01的值,其余的都是大于0.01的P-value值。
表5中,“null”代表这些模块没有富集到GO term上,观察可知只有模块272的GO分析三个方面的P-value值均小于0.01,说明三个方面富集效果较好的只有模块272,模块272与化学刺激参与嗅觉感知检测的功能、等离子体膜、电压门控离子通道的活性有关,从而说明该拓扑模块在生物功能上的意义比较明显,对人类疾病的影响比较大;但是在实际数据中该模块中包含的致病基因很少,我们推断出现这种现象是因为目前蛋白互作数据的缺失和与疾病相关的基因的不完整,因此可以根据272模块中的蛋白为现在未知疾病基因的预测提供理论支持。拓扑模块富集到GO上的比较少,而大部分P-value小于0.01,说明该拓扑模块在GO过程的三个方面的功能性特征基本关系一般,并不能代表模块在其相应的GO富集的功能特性,因而,这些拓扑模块在人类疾病中起不到关键的功能作用。
3.5较高与较低致病拓扑模块的功能比较
表6记录的是Ratio值大于1.2和Ratio值小于0.8的拓扑模块对应的GO术语的P-value小于0.01在其范围内的比例。比如,R>1.2时bp比例为0.877193指的是在R>1.2范围内的模块中GO分析的生物过程中P-value小于0.01的模块的数量占R>1.2的模块数量的比例。
从表6中可以看出,拓扑模块在R>1.2和R<0.8范围内,GO分析在生物过程bp、细胞组学cc、分子功能mf方面的富集比例有非常明显的差别。这种情况说明,较高致病拓扑模块的功能富集度比较低致病模块的功能富集度要高,同时也就说明了人类疾病与较高致病拓扑模块的功能具有较高的同质性。
4人类互作网络拓扑性质的功能同质性分析
现在的PPI数据和疾病基因数据只发现了大概10%左右,由于数据的不完整性和噪声数据的影响,所以在对疾病进行深层次研究时借助于疾病网络可以有效地帮助我们去探索人类疾病的分子生物机制。
目前有研究表明人类疾病的致病基因在PPI网络中并不是随机分布的,而是趋向于集中在某个连接相对紧密的局部模块中,也就是疾病模块,这也说明一旦发现了疾病的部分致病基因,那么从网络医学的角度就可以推论出现在已经发现的基因的邻居也有很大的可能是致病基因。
为了更好地理解疾病模块,对疾病模块的几个拓扑特性进行了分析,以便分析疾病模块的功能特性与拓扑特性之间的相互关系。本文主要使用了平均度、密度、平均最短路径、closeness中心性和betweenness中心性几个主要的网络属性应用于人类互作网络来探索疾病分类的功能特性与人类互作网络拓扑特性之间的相互关系。
4.1平均度
度是衡量网络中节点的一个重要属性,是指连接到某个节点的总的边数。在PPI网络中,度比较高的节点称为hub节点,是由疾病的必须基因进行编码的。平均度是衡量网络中节点与其他节点连接的程度的统计量。
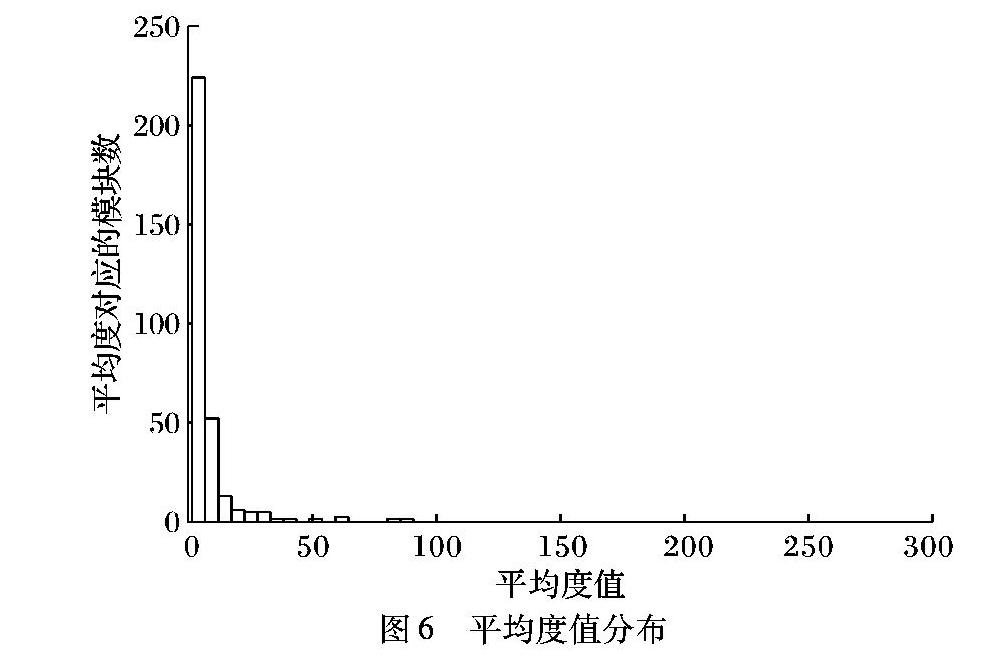
在人类互作网络划分出的314个模块中,平均度的值分布如图6所示。
图6中:横轴是平均度值,纵轴是平均度对应的模块度数。从图6中可以看出,平均度明显大于一般值的模块只有一个,其对应的为模块271,其他较大平均度值依次对应的是模块145、143、172、303、167,而其余大部分模块的平均度值都比较小,均小于50。这说明人类互作网络的拓扑模块中hub模块相对于普通模块少得多,并且在不同范围的介数中心性有明显的差别,总体呈幂律分布,体现了人类互作网络划分出的拓扑模块有多样性的特征。也说明不同的疾病模块、疾病分类在拓扑功能的表达中具有明显差异性,而hub模块271对于疾病的影响非常大,该模块可用于对多种疾病的研究。
4.2密度
网络的密度表示网络中节点间的边与同样节点数目的网络中的节点数的比例,体现的是网络的边密度。在人类互作网络划分出的314个模块中,密度的值分布如图7所示。
图7中:横坐标代表的是密度分布值,纵坐标代表的是坐标值为某一值的模块数,从图7中可看出,不同密度范围的模块数目具有多样性,大部分模块密度比较小,明显较大密度的拓扑模块不多。密度大于0.8的拓扑模块依次是模块34、144、271、39、205、227、19、260、286、311,而其余大部分模块的密度值都比较小,均小于0.50。这说明人类互作网络的拓扑模块中高密度模块相对于普通模块少得多,并且在不同范围的密度有明显的差别,总体分布不规律,体现了人类互作网络划分出的拓扑模块有多样性的特征。
4.3平均最短路径长度
平均最短路径描述了网络中节点间的平均分离程度,模块中的平均最短距离值越小说明该模块内的节点连接越紧密,模块所对应的子网的直径越小。划分出的314个模块的平均最短路径的值分布如图8所示。
图8中:横坐标代表的是平均最短路径长度值,纵坐标代表的是平均最短路径为某个值的模块数,
从图8中可看出,不同平均最短路径长度取值范围的模块数目具有多样性,大部分模块平均最短路径长度在(1.7,3.3)范围,模块平均最短路径长度明显较大和明显较小的拓扑模块并不多,总体呈正态分布。而网络中明显较小(小于1.5)的平均最短路径长度比网络中明显较大(大于3.5)的拓扑模块数量要多,说明网络中模块内的节点连接紧密的拓扑社团相对较多。
5结语
本文针对疾病所关联的拓扑模块功能同质性问题,采用MeSH疾病分类术语本体,采用比较成熟的聚类算法对来自整合的人类全局基因关系网络进行了了拓扑模块划分,并采用基因本体(GO)富集分析方法。最后通过比较分析发现,相较没有疾病相关的拓扑模块,具有显著疾病相关的拓扑模块在生物过程、细胞组分、分子功能等方面具有显著差异,从而为疾病的分子网络研究提供了重要启示。另外,对于疾病分类的拓扑模块的功能同质性分析,只进行了GO同质性分析,还可以进行更多的功能分析;同时对于网络的拓扑特性,可以将其结合相应的疾病分类、同质性结果等进行多角度的联合分析来共同辅助拓扑模块的功能同质性分析。
参考文献:
[1]BARABSI A-L, GULBAHCE N, LOSCALZO J. Network medicine: a network-based approach to human disease [J]. Nature Reviews Genetics, 2011, 12(1): 56-68.
[2]SHARAN R, ULITSKY I, SHAMIR R. Network-based prediction of protein function [J]. Molecular Systems Biology, 2007, 3(1): 88.
[3]LIN C-C, HSIANG J-T, WU C-Y, et al. Dynamic functional modules in co-expressed protein interaction networks of dilated cardiomyopathy [J]. BMC Systems Biology, 2010, 4(4): 138.
[4]ZHOU X, MENCHE J, BARABSI A-L, et al. Human symptoms-disease network [J]. Nature Communications, 2014, 5: 4212.
[5]BLONDEL V D, GUILLAUME J-L, LAMBIOTTE R, et al. Fast unfolding of communities in large networks [J]. Journal of Statistical Mechanics: Theory and Experiment, 2008, 2008: P10008
[6]NEWMAN M E, GIRVAN M. Finding and evaluating community structure in networks [J]. Physical Review E, 2004, 69(2): 026113.
[7]李乐,章毓晋.非负矩阵分解算法综述[J].电子学报,2008,36(4):737-743. (LI L, ZHANG Y J. A survey algorithms of non-negative matrix factorization [J]. Acta Electronica Sinica, 2008, 36(4): 737-743.)
[8]LEE D D, SEUNG H S. Learning the parts of objects by non-negative matrix factorization [J]. Nature, 1999, 401(6755): 788-791.
[9]CAI D, HE X, HAN J, et al. Graph regularized non-negative matrix factorization for data representation [J]. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2010, 33(8): 1548-1560.
[10]黄钢石,陆建江,张亚非.基于NMF的文本聚类方法[J].计算机工程,2004,30(11):113-114. (HUANG G S, LU J J, ZHANG Y F. Text clustering method based on non-negative matrix factorization[J].Computer Engineering, 2004, 30(11): 113-114.)
[11]YANG S, YE M. Multistability of α-divergence based NMF algorithms [J]. Computers & Mathematics with Applications, 2012, 64(2): 73-88.
