DNA Damage Response in Hematopoietic Stem Cell Ageing
2016-09-27TangliangLiZhongWeiZhouZhenyuJucZhaoQiWang
Tangliang Li*,Zhong-Wei Zhou,Zhenyu Juc,Zhao-Qi Wang
1Institute of Aging Research,School of Medicine,Hangzhou Normal University,Hangzhou 311121,China
2Leibniz Institute on Aging-Fritz Lipmann Institute(FLI),Jena D-07745,Germany
3Faculty of Biology and Pharmacy,Friedrich-Schiller University of Jena,Jena D-07745,Germany
REVIEW
DNA Damage Response in Hematopoietic Stem Cell Ageing
Tangliang Li1,*,a,Zhong-Wei Zhou2,b,Zhenyu Ju1,c,Zhao-Qi Wang2,3,d
1Institute of Aging Research,School of Medicine,Hangzhou Normal University,Hangzhou 311121,China
2Leibniz Institute on Aging-Fritz Lipmann Institute(FLI),Jena D-07745,Germany
3Faculty of Biology and Pharmacy,Friedrich-Schiller University of Jena,Jena D-07745,Germany
Available online 21 May 2016
Handled by Xingzhi Xu
KEYWORDS
Hematopoietic stem cells;
DNA damage response;
Epigenetics;
Ageing;
P53
AbstractMaintenance of tissue-specific stem cells is vital for organ homeostasis and organismal longevity.Hematopoietic stem cells(HSCs)are the most primitive cell type in the hematopoietic system.They divide asymmetrically and give rise to daughter cells with HSC identity(selfrenewal)and progenitor progenies(differentiation),which further proliferate and differentiate into full hematopoietic lineages.Mammalian ageing process is accompanied with abnormalities in the HSC self-renewal and differentiation.Transcriptional changes and epigenetic modulations have been implicated as the key regulators in HSC ageing process.The DNA damage response(DDR)in the cells involves an orchestrated signaling pathway,consisting of cell cycle regulation,cell death and senescence,transcriptional regulation,as well as chromatin remodeling.Recent studies employing DNA repair-deficient mouse models indicate that DDR could intrinsically and extrinsically regulate HSC maintenance and play important roles in tissue homeostasis of the hematopoietic system. In this review,we summarize the current understanding of how the DDR determines the HSC fates and finally contributes to organismal ageing.
Introduction
In adult animals,tissue homeostasis is maintained by a hierarchy of different types of cells,ranging from tissue-specific stem cells,progenitors,to somatic cells with different functions[1]. Stem cells are the most primitive cell population in a specific tissue,which on the one hand self-renew to sustain the stem cell pool,and on the other hand differentiate to generate their somatic progenies[1,2].Dysregulation of self-renewal and differentiation of tissue-specific stem cells compromises the stem cell function,resulting in loss of tissue maintenance and organismal ageing[1,3,4].
Among all types of tissue-specific stem cells,hematopoietic stem cell(HSC)is considered as the prototype to study the functions of genes of interest in adult stem cell self-renewal and maintenance,as well as their roles in physiological ageing[5,6].Under unperturbed conditions,HSCs reside within their niches(bone marrow stromal cells)and are exposed to systematic environments consisting of cytokine,chemokine(Figure 1),and other factors[7].The advantages of using the HSCs as the model to study stem cell ageing are mainly due to:(1)welldefined HSCs and their progenies with combinations of cell surface markers;(2)a panel of sophisticated in vitro assays to verify the HSC functions;and(3)the adoptive HSC transplantation assay as a gold standard to test stem cell functions[5].Using naturally-aged wild type mice and genetically-modified premature ageing mouse models[8-13],intrinsic and extrinsic factors contributing to the HSC ageing start to be unraveled[4,14-16]. Among them,cell cycle regulators,transcriptional factors,epigenetic modulators,and metabolic pathways have been implicated as important regulators for HSC self-renewal and maintenance during ageing process[10,12,17-23].
DNA lesions in cells originate from endogenous cellular activities,such as DNA replication and mitochondrial respiration,as well as exogenous stimuli,such as therapeutic drugs against cancers and medical exposure to irradiation,posing direct threats to the integrity of the cellular genetic information[24-26].If these DNA lesions could not be handled well,they will compromise cellular viability and drive the tumor formation[27,28].When it comes to the HSCs,improper repair of DNA lesions could negatively regulate the HSC maintenance and lead to HSC ageing[4,8,26].Here,we concisely discuss the signatures defining‘‘aged HSCs”and the role of genomic stability in HSC ageing.
Characteristics of HSCs in ageing hematopoietic system
Compared to the young individuals,the frequency(percentage of HSCs within bone marrows)and absolute numbers of HSCs,which are phenotypically designated with defined surface markers,increase in naturally-aged individuals of mice and humans(Figure 1)[8,29,30].However,HSCs in aged mice are defective in the self-renewal capacity[31].The adoptive bone marrow transplantation assay is the‘‘gold standard”to investigate the HSC functionality.Upon transplantation,HSCs are forced to enter the cell cycle and differentiate into different hematopoietic lineages[32].The sequential transplantation with the HSCs from the primary transplantation could be further employed to test the robustness of HSCs in self-renewal.During the serial transplantation,HSCs get exhausted and step into an‘‘aged”status[12,33].Using this serial adoptive transplantation assay,aged HSCs(HSCs from aged mice)showed limited repopulation ability to replenish the hematopoietic system in bone marrow-ablated congenic mice[12,29].The HSC transplantation assay indicates that the aged HSCs,in addition to a homing defect(a failure of transplanted donor HSCs trafficking to and engrafting in recipient bone marrows),only represent around 25%efficiency of HSCs from young animals[29].
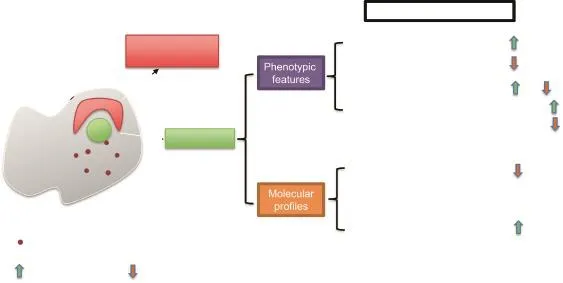
Figure 1 Characteristics of aged HSCs
Furthermore,aged HSCs have differentiation defects as well(Figure 1).Peripheral blood(PB)from aged mice contains a relative higher proportion of myeloid cells,such as Mac1+and Gr1+hematopoietic cells,as compared to the PB from young animals[29,34,35],which could be attributed to the higher proportion of myeloid progenitors generated in the bone marrow of aged mice[36].The biased myeloid hematopoiesis in the aged mice is detrimental to hematopoietic system functions since the dysregulated output of lymphoid and myeloid cells would compromise the immunological response upon injury or infection in the aged animals and further promote ageing.This skewed differentiation is cell-autonomous,since transplanting aged HSCs to young mice could recapitulate the phenotypes of‘‘ageing”hematopoietic compartments in these recipient mice[34,36]. The increased ratio of myeloid vs.lymphoid hematopoietic cells in ageing is further attributed to altered heterogeneity in HSC compartments during the ageing process[37,38]. Based on their differentiation capabilities,HSCs are further divided into lymphoid-biased HSCs(Ly-Bi HSCs),myeloidbiased HSCs(My-Bi HSCs),and balanced HSCs[17,37].The composition of HSC pools is shifted from Ly-Bi HSCs toward My-Bi HSCs during ageing.
In addition to the aforementioned phenotypically-defined characteristics,aged HSCs are distinct from young HSCs due totheiruniquetranscriptomicandepigenomicfeatures[39-41].Aged HSCs are implicated with marked increase in the expression of genes involved in stress responses,inflammation,and protein aggregation,while the expression of factors responsible for DDR and chromatin remodeling is reduced(Figure 1)[40].Accordingly,aged HSCs accumulate DNA lesions[8],are defective in protein homeostasis,and exhibit abnormal epigenetic landscapes on DNAs and histones[9,39,40].DNA methylation is enriched specifically on the promoter regions of lymphoid and erythroid lineage genes[9].On the contrary,promoters of genes responsible for the myeloid lineages exhibit reduced DNA methylation.This finding correlates with the skewed hematopoietic lineage output in aged mice[8,36,42].Furthermore,hypo-methylated cysteines and activechromatinmarkers,suchasH3K9me3and H3K27me3,are enriched in the promoter regions of genes in the Gene Ontology categories of cell adhesion,proliferation,and ribosome,which are expressed higher in the aged HSCs than in the young ones[39].Such transcriptional and epigenetic alterations could partially explain the phenotypic characteristics of aged HSCs,such as an increased mobilization,reduced homing ability,and loss of quiescence[29,39].
DNA damage accumulation in HSC ageing
In somatic cells,loss of genomic integrity compromises the cellular viability and threatens the genetic information passage from parent cells to daughter cells[43].Accumulation of genomic instability has been implicated in the hematopoietic malignancy,which could be derived from transformed HSCs[26,28]. DNA lesions initiate DDR and induce chromatin remodeling,epigenetic modification,as well as transcriptional regulation,which consequently activate a series of cellular responses including DNA repair,cell cycle checkpoint,cellular senescence,and cell death[24-26,43].All of these pan-genome,epigenome,and transcriptome modifications definitely generate systematic outcome to shape the dynamics of the HSCs in the context of self-renewal and differentiation[44,45].
The first direct link of DNA damage and HSC ageing comes from the analysis of double strand breakage(DSB)marker γ-H2AX in murine HSCs[8].Rossi and colleagues investigated the DNA damages inside aged murine HSCs and noticed that aged HSCs accumulate high levels of DNA DSBs[8]. However,a recent study suggested that those γ-H2AX marked‘‘DSB foci”may not be real DNA breaks.The‘‘DSB foci”are nucleolar-associated and represent the residual replication stress during the HSC cycling[46].The‘‘γ-H2AX foci”severs as the chromatin repressive marker for the silencing of rDNA transcription,which compromises the ribosome biogenesis in aged HSCs[46].Interestingly,Beerman et al.used the alkaline comet assay,which is extensively used in the field of DNA repair as the indicator for the DSBs and single strand breaks(SSBs)[47,48],to compare the DNA damages in young and aged quiescent HSCs.As a result,they noticed that aged HSCs have a high degree of DNA breaks,as indicated with increased‘‘Olive tail moment”[9,48].Similar to the murine HSCs,γ-H2AX antibody staining on human CD34+HSCs and hematopoietic progenitors reveals an significant accumulation of DSBs during normal ageing process[49].These data indicate that murine and human HSCs experience similar biological processes,namely genomic instability,during physiological ageing.
How are these DNA breaks generated in HSCs under physiological conditions during the ageing process?Reactive oxygen species(ROS)generated from metabolic pathways in quiescent HSCs and replication errors during HSC proliferation could be the threats to genome integrity in HSCs[33,50,51].Using different mouse models that harbor deficiencies in DNA repair pathways,it is found that loss of DNA repair factors results in accumulation of DNA damages in HSCs and severely-compromised capabilities of HSCs for self-renewal and differentiation under physiological conditions[8,11,52-54].For examples,knockout mice with defects in DNA DSB repair and quenching the ROS(such as Atm-/-mice)or in resolving the replication fork stalls(knockout of Fancd2 pathway members)are ageing-prone and show defective hematopoiesis[51,55,56].These findings strongly indicate that the proper repair of DNA damage is important for the maintenance of HSCs and protects against functional decline of HSCs during ageing[4,8,26,45].
DNA repair pathway choices in HSCs
In order to fix DNA breaks,cells are equipped with different repair mechanisms or repair factors[24,43].The choice of pathways to repair a DNA lesion is highly dependent on the cell cycle phase and/or the physiological status of a cell[25,57,58].Our knowledge of DNA repair in cell cycle stems from studies on the cycling somatic cells.Intriguingly,adults HSCs reside in quiescent status(G0)[14].Two repair scenarios have been proposed to repair ionizing radiation(IR)-generated DSBs in HSCs in G0 phase[59,60].Passegue´and her colleagues found that quiescent HSCs use the same repair program as in G1 phase of somatic cells,i.e.,non-homologous end joining(NHEJ)pathway to repair the IR-generated DNA lesions[60].In this scenario,upon DSB induction,protein complex comprising MRE11/RAD50/NBS1 is recruited to the DSB sites and activates ATM kinase,which phosphorylates MDC1/H2AX/53BP1/SMC1/KAP1 to alter the chromatin status around the DSBs and CHK2/p53 to initiate the cell cycle checkpoints and/or cell death signaling pathways[24,61-63].In addition,MRE11 nuclease resects the DNA strand at DSBs to generate micro-homology to facilitate the repair process[62,64,65].This repair pathway is considered as a low fidelity repair choice,since abnormal chromosome fusions,loss of genetic material around the breakage sites,and accumulation of genetic mutations could happen following the repair[63].Indeed,Passegue´and her colleagues found that 53BP1 foci(a marker of NHEJ)rather than RAD51(a marker of homologous recombination,HR)were prominently evident in IR-treated quiescent HSCs[60].Consequently,chromosome analysis with spectral karyotyping(SKY)on the hematopoietic progenitors derived from IR-irradiated HSCs reveals a great increase in genome instabilities,including chromosome fusions[60].However,this repair pathway could not effectively explain the accumulation of DNA break marker γ-H2AX in naturally-aged HSCs,because ligation of DNA breaks by NHEJ quenches the DDR signaling and therebygenerates γ-H2AX-free HSCs[8,42].It has been proposed that quiescence is a cellular status when HSC loses its stringent control of repair machineries[42,48].Beerman et al.conducted the in vitro short-term culture of isolated quiescent HSCs[48]. After 24 h,a significant reduction in γ-H2AX-marked DSBs was noticed when the HSCs enter the cell cycle[48],suggesting that proliferating HSCs repair DSB better.G0 HSCs apparently express low levels of DDR genes as compared to proliferating HSCs(such as fetal liver HSCs)and progenitors[48]. In this way,DNA damage signaling may be attenuated in quiescent HSCs,which is consistent with the previous finding on accumulation of DNA breaks in aged HSCs[8,42].
Once HSCs are mobilized and forced to enter cell cycle by in vivo administration of cytokine granulocyte-colony stimulating factor(G-CSF)or cultured in vitro in the presence of the stem cell factor(SCF),HSCs switch the repair mechanism from NHEJ toward HR[60].In S/G2/M HSCs,MRN complex recruits ATM and resects the DSBs to generate the single strand overhangs,which can activate ATR/CHK1 kinase[63,66].RAD51 is then loaded onto the exposed single strands and forms DNA/protein filaments to initiate strand invasion into their homologous chromosomes[67].As compared with NHEJ in G0/G1 cell cycle,HR is more stringent in keeping the genomic integrity.However,although HSCs can faithfully repair the DNA breaks when cycling[46,48,51],entry into cell cycle could be detrimental to the quality of HSCs because serial transplantation experiments in mice demonstrate that HSCs have limited replicative lifespan and multiple rounds of stress-induced HSC cycling can compromise self-renewal and differentiation capacity,leading to exhaustion of the HSC pool[12,46].
p53-p21/PUMA pathway in HSC cell fate determination
DNA damages exhaust HSCs in terms of self-renewal and differentiation by reducing the HSC pool(quantity)and compromising HSC stemness(quality)[26,35,45].Upon DNA damage,cells engage a serial of downstream cellular events including cell cycle arrest,apoptosis,and transcriptional reprogramming[43].Faithful DNA repair preserves the HSC genome integrity and sustains HSC stem cell identity in proper self-renewal and differentiation.However,depending on the repair efficiency for certain DNA lesions,HSCs undertake different fates toward permanent cell cycle arrest(senescence),cell death(HSC elimination)[68,69],and even differentiation[70-73].As the HSC fate determinant,the p53 pathway has been well studied in vivo[74,75].The p53 pathway is transiently activated after a single dose of IR or can be constantly activated in HSCs by persistent DNA damages,such as critically shortened telomeres[60,69,76].Downstream of p53 signaling,p53 trans-activates p21 to promote the cell survival by initiating cell cycle arrest for DNA repair or cellular senescence,while induced expression of the p53 upregulated modulator of apoptosis(PUMA)by p53 is responsible for cellular clearance(Figure 2)[13,68].Inhibition of either branch of p53-p21 or p53-PUMA benefits HSC selfrenewal and maintenance in several cases of HSC ageing mice models[74,77-79].Complete p53 loss renders cytoprotective effects on IR-damaged HSCs[80]and promotes symmetric division of HSCs to expand the HSC pool[74,81,82]. However,these p53-null HSCs show defective differentiation and are tumor-prone,indicating that p53 null compromises the HSC quality[74,78].In this regard,the balance of p21 and PUMA downstream of p53 signaling is essential for the maintenance of HSCs and hematopoietic system[75,83].
Compared to hematopoietic progenitors,murine HSCs are resistant to acute DNA damage induction and prone to survival[60].This is likely due to a high expression level of the pro-survival genes in murine HSCs[60].In response to acute DNA damage,HSCs tend to be arrested and reside in the senescent status,suggesting that p53-p21 branch is activated in HSCs preferably to limit HSC self-renewal[77,84].Loss of p21 in the mouse model with persistent DNA damage(the 3rd generation of Terc-/-mice;G3 Terc-/-)with criticallyshortened telomeres could partially rescue ageing phenotypes by improving the repopulation capacity and self-renewal of HSCs[77].Furthermore,activated p53-PUMA branch is responsible for the HSC death upon lethal dose of γirradiation(10 Gy),since loss of PUMA protects the HSCs and extends the lifespan of irradiated mice[85].The constitutive activation of p53 signaling in mice lines expressing p53 phosphorylation mutations(T21D and S23D),a C-terminal truncatedp53allele,orothergenemutationsconfers premature ageing of hematopoietic systems[13,86,87].Genetic ablation of Puma restores the viability of HSCs,indicating p53-PUMA limiting the HSC pool size[85,86].These data point to a promising therapeutic strategy to protect HSCs and prolong healthy lifespan with p21 or PUMA inhibitors. However,it is of note that p21-null HSCs exhibit selfrenewaldefectsinserialtransplantationassay,while Puma-null HSCs are superior to their wild type controls[86,88].These findings indicate that p53-p21 and p53-PUMA have differential roles in mediating HSC fates and ageing due to different extents of DNA lesions(Figure 2).
Persistent DNA damage creates a pro-ageing environment for HSCs
Intrinsic defects in repairing DNA damages and their contributionstocompromisedHSCself-renewalandageing process have been extensively studied in recent years.However,ageing environments,such as mis-regulated cytokine factor secretion and altered stem cells niches,can all affect HSC maintenance(Figure 1)[4,76,89,90].An interesting example of the environmental impact on the HSC selfrenewal and differentiation comes from the parabiosis assay by surgical connection between young and aged mice with the circulatory blood system[91,92].In this assay,young and aged HSCs are unanimously exposed to a common systematic environment,such as serum factors and osteoblast niches.Multi-organ analysis shows that the interconnection of young and aged mice significantly improves tissue homeostasis including HSCs and hematopoietic system of the aged mice.These data strongly indicate that a young systematic environment can rejuvenate the aged HSCs[91,92]. Although the search for the key factors in the systematic environment that contribute to the ageing and rejuvenation of stem cells is still ongoing,these findings conceptually prove that the HSCs,in addition to the intrinsic regulation,could be functionally modulated by the environmental cues. On the other hand,parabiosis assay highlights a novelconcept that HSC ageing could be delayed or partially reversed by rejuvenating serum factors[69,91,92].

Figure 2 p53 signaling in HSC fate determination toward ageing
Does DNA damage generate a systematic change and promote the HSC ageing?The analysis of HSCs from G3 Terc-/-mice unveils some hints on this question.The criticallyshortened telomeres in G3 Terc-/-mice can be recognized as persistent DNA breaks,which constantly activate the p53-p21/PUMA pathway in tissue-specific stem cells and their somatic progenies[77,89,93].Ju et al.analyzed the interplay between HSCs and their niches,i.e.,mesenchymal stem cell(MSC)-derived bone marrow stromal cells.In G3 Terc-/-mice,the functionality of both MSCs and bone marrow stromal cells is compromised.In addition,the high level of G-CSF cytokine in the G3 Terc-/-mice serum significantly reduces the engraftment of HSCs in bone marrow niches[76].G-CSF inhibition leads to the improved engraftment and functionality of HSCs.These findings suggest that a cellular response from those‘‘damaged”cells with persistent DNA lesions caused by telomere shortening could alter the local environment(such as HSC niches)or systematic environment(i.e.,cytokines or chemokines in serum)and confer a deleterious effect on self-renewal and maintenance of HSCs[69,76,89].
How persistent DNA damage signaling can change the systematic environment?One possibility could be the senescenceassociated secretory phenotype(SASP)[94,95].Campisi and colleagues found that senescent cells,although permanently arrested in cell cycle,is metabolically active in producing inflammatory factors(SASP cytokines),such as IL-6,IL-10,INF γ,and G-CSF[94].Furthermore,not only senescent cells,but also cells with persistent DNA damages,exhibit SASP and secrete pro-inflammatory cytokines to change systematic environment in the animal tissues[96].Activated NF-κB signaling has been implicated in SASP,since inhibition of NF-κB signaling by knocking down of p65 greatly alleviates the expression ofSASPcytokines[97-99].Furthermore,activationof p38MAPK kinase activity by various stimuli promotes SASP induction.p38MAPK sits upstream of NF-κB signaling and regulates the NF-κB activity.Interestingly,although p53 is not required for initiating SASP,p53 restrains SASP once the cellular senescence is established,since p53-null cells show enhanced expression of SASP cytokines[99].The inhibitory effects of SASP by p53 could be attributed to the fact that p53 restrains p38MAPK activity via its DDR-independent activity.In this sense,p53 DDR-independent signaling may provide protective roles in maintaining HSC homeostasis by inhibiting SASP in the systematic environment of hematopoietic system(Figure 2).
The secreted inflammatory factors from these cells with persistent DNA damages,may be detrimental to the HSC self-renewal and maintenance.For example,G-CSF mobilizes the HSCs out of bone marrow niches and impairs their engraftment[76,100].IL6,INFα,and INF γ have been implicated inpromoting the cell cycle entry of HSCs,resulting in HSC exhaustion toward ageing[90,101].Tissues in aged animals are enriched in senescent somatic cells that contain persistent DNAdamages.Furthermore,ageingprocesspositively correlates with increased inflammatory responses[102],which may further ameliorate the HSC maintenance and promote ageing[103].Recent studies indicate that interfering with SASP by blocking the cytokine production pathway or eliminating cytokine-producing cells in tissues greatly improves the tissue function and animal lifespan.Clearance of p16Ink4a-positive senescent cells in progeroid BubR1 mutant mice could substantially delay the onset of ageing phenotypes and even rejuvenate the ageing tissues when such cellular clearance was applied in late-life of BubR1 mutants[104].Furthermore,Chang et al. employed ABT263,a specific chemical inhibitor for Bcl-2 and Bcl-xL,to induce the apoptosis of senescent HSCs.They found that ABT263 treatment restores the functionality of HSCs in the sub-lethally irradiated wild type mice and naturally-aged mice,thus greatly improving the healthy lifespan of mice[105].The data further confirm the assumption that senescent cells with persistent DNA damage can establish a systematic environment driving HSC ageing.
Conclusions and perspectives
Integrities of HSC pool and HSC quality are considered as the key factors contributing to the organismal ageing in mammals[4,29].DDR plays essential roles in the maintenance of these two HSC features[8,44,45,52,68].Deficiency in DNA repair results in the accumulation of unrepaired DNA breaks in aged HSCs,while persistent DNA breaks in hematopoietic cells and HSC niches create a pro-inflammatory environment to promote HSC entry into cell cycle and proliferation,which consequently exhausts HSCs[69].It would be very tricky to experimentallymodifythoseageingHSCswithgenetic approaches in order to achieve the better DNA repair and functional improvement.Instead,using bio-active cytokines to interfere with the systematic ageing environment or using small chemical molecules to specially remove the‘‘aged”HSCs would be the reliable and practical strategies to rejuvenate theageingHSCsandprolongthehealthylifespanof mammals[105,106].
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
TL is currently supported by the National Natural Science Foundation of China(Grant No.81571380),and the Natural Science Foundation of Zhejiang Province-China(Grant No. LY16H080009).ZJ is supported by the National Natural ScienceFoundationofChina(GrantNos.81130074,81420108017,and 81525010).TL and ZJ are both funded by the National Key R&D Plan from the Ministry of Science and Technology of China(Grant No.SQ2016ZY05002341). ZQW is partially supported by the Deutsche Forschungsgemeinschaft(DFG),Germany.
References
[1]Weissman IL.Stem cells:units of development,units of regeneration,and units in evolution.Cell 2000;100:157-68.
[2]He S,Nakada D,Morrison SJ.Mechanisms of stem cell selfrenewal.Annu Rev Cell Dev Biol 2009;25:377-406.
[3]Signer RA,Morrison SJ.Mechanisms that regulate stem cell aging and life span.Cell Stem Cell 2013;12:152-65.
[4]Lopez-Otin C,Blasco MA,Partridge L,Serrano M,Kroemer G. The hallmarks of aging.Cell 2013;153:1194-217.
[5]Orkin SH,Zon LI.Hematopoiesis:an evolving paradigm for stem cell biology.Cell 2008;132:631-44.
[6]Mikkola HK,Orkin SH.The journey of developing hematopoietic stem cells.Development 2006;133:3733-44.
[7]Morrison SJ,Scadden DT.The bone marrow niche for haematopoietic stem cells.Nature 2014;505:327-34.
[8]Rossi DJ,Bryder D,Seita J,Nussenzweig A,Hoeijmakers J,Weissman IL.Deficiencies in DNA damage repair limit the functionofhaematopoieticstemcellswithage.Nature 2007;447:725-9.
[9]Beerman I,Bock C,Garrison BS,Smith ZD,Gu H,Meissner A,et al.Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging.Cell Stem Cell 2013;12:413-25.
[10]Challen GA,Sun D,Mayle A,Jeong M,Luo M,Rodriguez B,et al.Dnmt3a and Dnmt3b have overlapping and distinct functionsinhematopoieticstemcells.CellStemCell 2014;15:350-64.
[11]Zhang S,Yajima H,Huynh H,Zheng J,Callen E,Chen HT,et al.Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair.J Cell Biol 2011;193:295-305.
[12]Janzen V,Forkert R,Fleming HE,Saito Y,Waring MT,Dombkowski DM,et al.Stem-cell ageing modified by the cyclindependent kinase inhibitor p16INK4a.Nature 2006;443:421-6.
[13]Dumble M,Moore L,Chambers SM,Geiger H,Van Zant G,Goodell MA,et al.The impact of altered p53 dosage on hematopoieticstemcelldynamicsduringaging.Blood 2007;109:1736-42.
[14]Chen J,Astle CM,Harrison DE.Genetic regulation of primitive hematopoieticstemcellsenescence.ExpHematol 2000;28:442-50.
[15]Oshima M,Iwama A.Epigenetics of hematopoietic stem cell aging and disease.Int J Hematol 2014;100:326-34.
[16]Rossi DJ,Jamieson CH,Weissman IL.Stems cells and the pathways to aging and cancer.Cell 2008;132:681-96.
[17]Challen GA,Boles NC,Chambers SM,Goodell MA.Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1.Cell Stem Cell 2010;6:265-78.
[18]Luo M,Jeong M,Sun D,Park HJ,Rodriguez BA,Xia Z,et al. Long non-coding RNAs control hematopoietic stem cell function.Cell Stem Cell 2015;16:426-38.
[19]Zhao Y,Zhou J,Liu D,Dong F,Cheng H,Wang W,et al. ATF4 plays a pivotal role in the development of functional hematopoieticstemcellsinmousefetalliver.Blood 2015;126:2383-91.
[20]Wang H,Diao D,Shi Z,Zhu X,Gao Y,Gao S,et al.SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of Wnt signaling.Cell Stem Cell 2016;18:495-507.
[21]Wang X,Chu Y,Wang W,Yuan W.MTORC signaling in hematopoiesis.Int J Hematol 2016;103:510-8.
[22]Qian P,He XC,Paulson A,Li Z,Tao F,Perry JM,et al.The Dlk1-Gtl2 locus preserves LT-HSC function by inhibiting the PI3K-mTOR pathway to restrict mitochondrial metabolism.Cell Stem Cell 2016;18:214-28.
[23]Lee JY,Nakada D,Yilmaz OH,Tothova Z,Joseph NM,Lim MS,et al.MTOR activation induces tumor suppressors thatinhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion.Cell Stem Cell 2010;7:593-605.
[24]D’Amours D,Jackson SP.The Mre11 complex:at the crossroads of DNA repair and checkpoint signalling.Nat Rev Mol Cell Biol 2002;3:317-27.
[25]Branzei D,Foiani M.Regulation of DNA repair throughout the cell cycle.Nat Rev Mol Cell Biol 2008;9:297-308.
[26]Kenyon J,Gerson SL.The role of DNA damage repair in aging of adult stem cells.Nucleic Acids Res 2007;35:7557-65.
[27]Hoeijmakers JH.Genome maintenance mechanisms for preventing cancer.Nature 2001;411:366-74.
[28]Hoeijmakers JH.Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech Ageing Dev 2007;128:460-2.
[29]Geiger H,Denkinger M,Schirmbeck R.Hematopoietic stem cell aging.Curr Opin Immunol 2014;29:86-92.
[30]Pang WW,Price EA,Sahoo D,Beerman I,Maloney WJ,Rossi DJ,et al.Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age.Proc Natl Acad Sci U S A 2011;108:20012-7.
[31]Roobrouck VD,Ulloa-Montoya F,Verfaillie CM.Self-renewal and differentiation capacity of young and aged stem cells.Exp Cell Res 2008;314:1937-44.
[32]Kondo M,Wagers AJ,Manz MG,Prohaska SS,Scherer DC,Beilhack GF,et al.Biology of hematopoietic stem cells and progenitors:implications for clinical application.Annu Rev Immunol 2003;21:759-806.
[33]Kamminga LM,van Os R,Ausema A,Noach EJ,Weersing E,Dontje B,et al.Impaired hematopoietic stem cell functioning after serial transplantation and during normal aging.Stem Cells 2005;23:82-92.
[34]Sudo K,Ema H,Morita Y,Nakauchi H.Age-associated characteristics of murine hematopoietic stem cells.J Exp Med 2000;192:1273-80.
[35]RossiDJ,BryderD,WeissmanIL.Hematopoieticstemcellaging: mechanism and consequence.Exp Gerontol 2007;42:385-90.
[36]Rossi DJ,Bryder D,Zahn JM,Ahlenius H,Sonu R,Wagers AJ,et al.Cell intrinsic alterations underlie hematopoietic stem cell aging.Proc Natl Acad Sci U S A 2005;102:9194-9.
[37]Beerman I,Bhattacharya D,Zandi S,Sigvardsson M,Weissman IL,Bryder D,et al.Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion.Proc Natl Acad Sci U S A 2010;107:5465-70.
[38]Muller-Sieburg CE,Sieburg HB,Bernitz JM,Cattarossi G.Stem cell heterogeneity:implications for aging and regenerative medicine.Blood 2012;119:3900-7.
[39]Sun D,Luo M,Jeong M,Rodriguez B,Xia Z,Hannah R,et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal.Cell Stem Cell 2014;14:673-88.
[40]Chambers SM,Shaw CA,Gatza C,Fisk CJ,Donehower LA,Goodell MA.Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation.PLoS Biol 2007;5:e201.
[41]Sinclair DA,Oberdoerffer P.The ageing epigenome:damaged beyond repair?Ageing Res Rev 2009;8:189-98.
[42]Rossi DJ,Seita J,Czechowicz A,Bhattacharya D,Bryder D,Weissman IL.Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging.Cell Cycle 2007;6:2371-6.
[43]Jackson SP,Bartek J.The DNA-damage response in human biology and disease.Nature 2009;461:1071-8.
[44]Mandal PK,Blanpain C,Rossi DJ.DNA damage response in adult stem cells:pathways and consequences.Nat Rev Mol Cell Biol 2011;12:198-202.
[45]Blanpain C,Mohrin M,Sotiropoulou PA,Passegue E.DNA-damage response in tissue-specific and cancer stem cells.Cell Stem Cell 2011;8:16-29.
[46]Flach J,Bakker ST,Mohrin M,Conroy PC,Pietras EM,Reynaud D,et al.Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells.Nature 2014;512:198-202.
[47]Klaude M,Eriksson S,Nygren J,Ahnstrom G.The comet assay: mechanismsandtechnicalconsiderations.MutatRes 1996;363:89-96.
[48]Beerman I,Seita J,Inlay MA,Weissman IL,Rossi DJ.Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle.Cell Stem Cell 2014;15:37-50.
[49]Rube CE,Fricke A,Widmann TA,Furst T,Madry H,Pfreundschuh M,et al.Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One 2011;6:e17487.
[50]Naka K,Muraguchi T,Hoshii T,Hirao A.Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells.Antioxid Redox Signal 2008;10:1883-94.
[51]Walter D,Lier A,Geiselhart A,Thalheimer FB,Huntscha S,Sobotta MC,et al.Exit from dormancy provokes DNA-damageinducedattritioninhaematopoieticstemcells.Nature 2015;520:549-52.
[52]Nijnik A,Woodbine L,Marchetti C,Dawson S,Lambe T,Liu C,et al.DNA repair is limiting for haematopoietic stem cells during ageing.Nature 2007;447:686-90.
[53]Bender CF,Sikes ML,Sullivan R,Huye LE,Le Beau MM,Roth DB,et al.Cancer predisposition and hematopoietic failure in Rad50(S/S)mice.Genes Dev 2002;16:2237-51.
[54]Parmar K,Kim J,Sykes SM,Shimamura A,Stuckert P,Zhu K,et al.Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1.Stem Cells 2010;28:1186-95.
[55]Ito K,Hirao A,Arai F,Takubo K,Matsuoka S,Miyamoto K,et al.Reactive oxygen species act through p38 MAPK to limit thelifespanofhematopoieticstemcells.NatMed 2006;12:446-51.
[56]Ito K,Hirao A,Arai F,Matsuoka S,Takubo K,Hamaguchi I,et al.Regulation of oxidative stress by ATM is required for selfrenewal of haematopoietic stem cells.Nature 2004;431:997-1002.
[57]Zhao B,Zhang WD,Duan YL,Lu YQ,Cun YX,Li CH,et al. Filia is an ESC-specific regulator of DNA damage response and safeguards genomic stability.Cell Stem Cell 2015;16:684-98.
[58]Ahuja AK,Jodkowska K,Teloni F,Bizard AH,Zellweger R,Herrador R,et al.A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells.Nat Commun 2016;7:10660.
[59]Ambrosio S,Di Palo G,Napolitano G,Amente S,Dellino GI,Faretta M,et al.Cell cycle-dependent resolution of DNA double-strand breaks.Oncotarget 2016;7:4949-60.
[60]Mohrin M,Bourke E,Alexander D,Warr MR,Barry-Holson K,Le Beau MM,et al.Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis.Cell Stem Cell 2010;7:174-85.
[61]Bruhn C,Zhou ZW,Ai H,Wang ZQ.The essential function of the MRN complex in the resolution of endogenous replication intermediates.Cell Rep 2014;6:182-95.
[62]Saidi A,Li T,Weih F,Concannon P,Wang ZQ.Dual functions of Nbs1 in the repair of DNA breaks and proliferation ensure proper V(D)J recombination and T-cell development.Mol Cell Biol 2010;30:5572-81.
[63]Yang YG,Saidi A,Frappart PO,Min W,Barrucand C,Dumon-Jones V,et al.Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA doublestrand breaks.EMBO J 2006;25:5527-38.
[64]Symington LS.End resection at double-strand breaks:mechanism and regulation,vol.6.Cold Spring Harb Perspect Biol;2014.
[65]Xie A,Kwok A,Scully R.Role of mammalian Mre11 in classical and alternative nonhomologous end joining.Nat Struct Mol Biol 2009;16:814-8.
[66]Jazayeri A,Falck J,Lukas C,Bartek J,Smith GC,Lukas J,et al. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks.Nat Cell Biol 2006;8:37-45.
[67]Baumann P,West SC.Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci 1998;23:247-51.
[68]Sperka T,Wang J,Rudolph KL.DNA damage checkpoints in stemcells,ageingandcancer.NatRevMolCellBiol 2012;13:579-90.
[69]Behrens A,van Deursen JM,Rudolph KL,Schumacher B. Impact of genomic damage and ageing on stem cell function.Nat Cell Biol 2014;16:201-7.
[70]Weiss CN,Ito K.DNA damage:a sensible mediator of the differentiation decision in hematopoietic stem cells and in leukemia.Int J Mol Sci 2015;16:6183-201.
[71]Inomata K,Aoto T,Binh NT,Okamoto N,Tanimura S,Wakayama T,et al.Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation.Cell 2009;137:1088-99.
[72]Sherman MH,Bassing CH,Teitell MA.Regulation of cell differentiation by the DNA damage response.Trends Cell Biol 2011;21:312-9.
[73]Santos MA,Faryabi RB,Ergen AV,Day AM,Malhowski A,Canela A,et al.DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier.Nature 2014;514:107-11.
[74]Liu Y,Elf SE,Miyata Y,Sashida G,Liu Y,Huang G,et al.P53 regulates hematopoietic stem cell quiescence.Cell Stem Cell 2009;4:37-48.
[75]Liu Y,Elf SE,Asai T,Miyata Y,Liu Y,Sashida G,et al.The p53tumorsuppressorproteinisacriticalregulatorof hematopoietic stem cell behavior.Cell Cycle 2009;8:3120-4.
[76]Ju Z,Jiang H,Jaworski M,Rathinam C,Gompf A,Klein C,et al.Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment.Nat Med 2007;13:742-7.
[77]Sperka T,Song Z,Morita Y,Nalapareddy K,Guachalla LM,Lechel A,et al.Puma and p21 represent cooperating checkpoints limiting self-renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction.Nat Cell Biol 2012;14:73-9.
[78]Begus-Nahrmann Y,Lechel A,Obenauf AC,Nalapareddy K,Peit E,Hoffmann E,et al.P53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice.Nat Genet 2009;41:1138-43.
[79]Choudhury AR,Ju Z,Djojosubroto MW,Schienke A,Lechel A,Schaetzlein S,et al.Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation.Nat Genet 2007;39:99-105.
[80]Marusyk A,Porter CC,Zaberezhnyy V,DeGregori J.Irradiation selects for p53-deficient hematopoietic progenitors.PLoS Biol 2010;8:e1000324.
[81]Cicalese A,Bonizzi G,Pasi CE,Faretta M,Ronzoni S,Giulini B,etal.Thetumorsuppressorp53regulatespolarityofself-renewing divisions in mammary stem cells.Cell 2009;138:1083-95.
[82]Insinga A,Cicalese A,Faretta M,Gallo B,Albano L,Ronzoni S,et al.DNA damage in stem cells activates p21,inhibits p53,and induces symmetric self-renewing divisions.Proc Natl Acad Sci U S A 2013;110:3931-6.
[83]Bonizzi G,Cicalese A,Insinga A,Pelicci PG.The emerging role of p53 in stem cells.Trends Mol Med 2012;18:6-12.
[84]Wang Y,Schulte BA,LaRue AC,Ogawa M,Zhou D.Total body irradiation selectively induces murine hematopoietic stem cell senescence.Blood 2006;107:358-66.
[85]Yu H,Shen H,Yuan Y,XuFeng R,Hu X,Garrison SP,et al. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose gamma-irradiation. Blood 2010;115:3472-80.
[86]Liu D,Ou L,Clemenson Jr GD,Chao C,Lutske ME,Zambetti GP,et al.Puma is required for p53-induced depletion of adult stem cells.Nat Cell Biol 2010;12:993-8.
[87]Belle JI,Langlais D,Petrov JC,Pardo M,Jones RG,Gros P,et al.P53 mediates loss of hematopoietic stem cell function and lymphopenia in Mysm1 deficiency.Blood 2015;125:2344-8.
[88]Cheng T,Rodrigues N,Shen H,Yang Y,Dombkowski D,Sykes M,et al.Hematopoietic stem cell quiescence maintained by p21cip1/waf1.Science 2000;287:1804-8.
[89]Song Z,Ju Z,Rudolph KL.Cell intrinsic and extrinsic mechanisms of stem cell aging depend on telomere status.Exp Gerontol 2009;44:75-82.
[90]Baldridge MT,King KY,Goodell MA.Inflammatory signals regulatehematopoieticstemcells.TrendsImmunol2011;32:57-65.
[91]Conboy IM,Conboy MJ,Wagers AJ,Girma ER,Weissman IL,Rando TA.Rejuvenation of aged progenitor cells by exposure to a young systemic environment.Nature 2005;433:760-4.
[92]Conboy MJ,Conboy IM,Rando TA.Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity.Aging Cell 2013;12:525-30.
[93]SchaetzleinS,KodandaramireddyNR,JuZ,LechelA,Stepczynska A,Lilli DR,et al.Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomeredysfunctional mice.Cell 2007;130:863-77.
[94]Coppe JP,Patil CK,Rodier F,Sun Y,Munoz DP,Goldstein J,et al.Senescence-associated secretory phenotypes reveal cellnonautonomous functions of oncogenic RAS and the p53 tumor suppressor.PLoS Biol 2008;6:2853-68.
[95]Coppe JP,Desprez PY,Krtolica A,Campisi J.The senescenceassociated secretory phenotype:the dark side of tumor suppression.Annu Rev Pathol 2010;5:99-118.
[96]Rodier F,Coppe JP,Patil CK,Hoeijmakers WA,Munoz DP,Raza SR,et al.Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion.Nat Cell Biol 2009;11:973-9.
[97]Salminen A,Kauppinen A,Kaarniranta K.Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype(SASP).Cell Signal 2012;24:835-45.
[98]Chien Y,Scuoppo C,Wang X,Fang X,Balgley B,Bolden JE,et al.Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity.Genes Dev 2011;25:2125-36.
[99]Freund A,Patil CK,Campisi J.P38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype.EMBO J 2011;30:1536-48.
[100]Tesio M,Oser GM,Baccelli I,Blanco-Bose W,Wu H,Gothert JR,et al.Pten loss in the bone marrow leads to G-CSF-mediated HSC mobilization.J Exp Med 2013;210:2337-49.
[101]Baldridge MT,King KY,Boles NC,Weksberg DC,Goodell MA.Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection.Nature 2010;465:793-7.
[102]Franceschi C,Bonafe M,Valensin S,Olivieri F,De Luca M,Ottaviani E,et al.Inflamm-aging.An evolutionary perspective on immunosenescence.Ann N Y Acad Sci 2000;908:244-54.
[103]King KY,Goodell MA.Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response.Nat Rev Immunol 2011;11:685-92.
[104]Baker DJ,Wijshake T,Tchkonia T,LeBrasseur NK,Childs BG,van de Sluis B,et al.Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders.Nature 2011;479:232-6.
[105]Chang J,Wang Y,Shao L,Laberge RM,Demaria M,Campisi J,et al.Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice.Nat Med 2016;22:78-83.
[106]Sinha M,Jang YC,Oh J,Khong D,Wu EY,Manohar R,et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle.Science 2014;344:649-52.
29 February 2016;revised 20 April 2016;accepted 24 April 2016
*Corresponding author.
E-mail:li.tangliang@hznu.edu.cn(Li T).aORCID:0000-0003-0671-9166.bORCID:0000-0002-5936-9094.cORCID:0000-0002-9525-6521.dORCID:0000-0002-8336-3485.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.04.002
1672-0229ⓒ2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Maintenance of Genome Stability
- Endogenous DNA Damage and Repair Enzymes—A short summary of the scientific achievements of Tomas Lindahl,Nobel Laureate in Chemistry 2015
- Connecting Malfunctioning Glial Cells and Brain Degenerative Disorders
- Topoisomerase I in Human Disease Pathogenesis and Treatments
- New Edges of RNA Adenosine Methylation Modifications