乳酸菌基因敲除技术的研究进展
2016-09-26杜胜阳王斌斌冯佳侯斌晓乔建军
杜胜阳,王斌斌,冯佳,侯斌晓,乔建军
(天津大学 化工学院制药工程系,系统生物工程教育部重点实验室,天津,300072)
乳酸菌基因敲除技术的研究进展
杜胜阳,王斌斌,冯佳,侯斌晓,乔建军*
(天津大学 化工学院制药工程系,系统生物工程教育部重点实验室,天津,300072)
乳酸菌是革兰氏阳性菌,广泛用于食品、医药、轻工业及饲料工业等行业。对乳酸菌进行遗传操作是调节、优化其代谢途径的基础。通过基因敲除技术改造乳酸菌基因组,可以判断基因组中未知基因所编码产物的功能,有目的、有选择地使乳酸菌生产有益的异源基因产物。基因敲除技术已成为当前乳酸菌分子生物学研究的热点。文中对几种乳酸菌基因敲除策略研究进展及影响敲除效率的因素进行了综述,分析存在的问题并初步探讨展望了乳酸菌敲除技术的未来发展趋势。
基因敲除;乳酸菌;同源重组;单链重组;敲除效率
乳酸菌(lactic acid bacteria, LAB)是一类能发酵碳水化合物且主要产物为乳酸的无芽孢、革兰氏阳性细菌的通称。目前至少分为18个属,包括乳杆菌属(Lactobacillus)、乳球菌属(Lactococcus)、链球菌属(Streptococcus)、肠球菌属(Enterococcus)、明串珠菌属(Leuconostoc)、双歧杆菌属(Bifidobacterium)等。乳酸菌代谢相对简单,具有很好的生物安全性,是公认的安全级微生物(generally recognized as safe, GRAS),被广泛用于食品、医药、轻工业及饲料工业等行业[1]。
随着分子生物学的发展,基因测序技术的改革,以及后基因组时代的到来,人们已经完成了对多种乳酸菌基因组的测序,从基因水平上更加清楚地认识、了解它的代谢规律。乳酸菌基因组中含有大量非必需基因,包括次级代谢产物基因簇、插入序列、冗余序列等。敲除这些非必需基因,可以改造得到最小化基因组的乳酸菌。
随着合成生物学的发展,微生物基因组简化成为研究热点[2]。乳酸菌的基因组简化后,能优化其代谢网络,减少副产物,提高了对底物和能量的利用效率,提升代谢网络的可控制性以及产物的可预测性。目前,基因组简化的研究在大肠杆菌、谷氨酸棒杆菌、枯草芽孢杆菌、酿酒酵母等模式微生物中已相继被报道[3-5]。其中,对大肠杆菌基因敲除系统的研究比较成熟,如高效的Red/ET重组系统。然而基因组简化的研究在乳酸菌中尚不成熟,更高效的基因敲除系统有待探究。目前,大部分乳酸菌基因敲除技术还是依赖于传统的同源重组双交换方法。本文就近年来乳酸菌基因敲除技术取得的进展进行综述,并探讨了影响敲除效率的有关因素,旨在为今后的深入研究提供有价值的参考。
1 乳酸菌基因敲除原理概述
基因敲除技术是20世纪80年代发展起来的一项重要的分子生物学技术,其原理是利用特定方法使目标基因失活或者缺失,对染色体进行定点修饰改造,从而改变细胞的遗传特性。运用基因敲除技术,可以改变乳酸菌的代谢流向,阻断不必要次级代谢产物的积累,降低发酵成本,大幅度提高目的产物的生产水平及纯度。另外,基因敲除技术还可用于乳酸菌基因结构与功能研究[6]。
经典基因敲除技术的理论依据是同源重组。同源重组依赖于基因同源序列的联会,DNA分子间或者分子内交换对等的部分,发生重新组合。根据同源重组原理,合理设计并通过酶切连接、融合PCR构建不同结构的重组载体,然后将其导入宿主细胞中,实现对宿主染色体DNA序列的插入、缺失或者置换突变。乳酸菌中的同源重组是个复杂的过程,通常分为三个阶段,即前联会体阶段、联会体形成阶段和Holiday 结构(Holiday Juncture Structure)的拆分阶段,期间需要RecA、RecBCD、RecF、RecR、RuvAB等重要蛋白参与。
另外,还可以依据位点特异性重组对乳酸菌进行基因敲除。它的原理是在重组酶催化下,含有特异位点的两条DNA链会发生重组。依据特异位点的序列和排列方向,重组以后可能出现3种结果:整合(integration)、切除(excision)、倒位(inversion)[7]。
位点特异性重组酶主要包括两大家族,分别为整合酶系和解离酶系。整合酶家族有两个代表性的重组酶系统分别是Cre/loxP、FLP/FRT重组酶系统。解离酶家族中,常用于乳酸菌的重组酶是lactococcal噬菌体的TP901-1整合酶,它能够特异性介导attP位点与attB位点间的重组反应。
2 乳酸菌基因敲除策略
乳酸菌中的基因敲除大致可以分为3类:同源单双交换基因敲除、位点特异性重组、线形DNA重组基因敲除。
2.1利用同源重组单交换双交换进行敲除
同源重组单双交换的首要条件是构建重组载体,运用PCR将目标基因上游以及下游片段或者不完整的靶基因扩增出来,连接到敲除载体上,然后转化进入宿主细胞,发生一次或两次同源重组,以敲除或者破坏基因。
在对目标基因进行敲除过程中,只发生一次同源重组,称为同源单交换,如图1所示。单交换操作简单、易行,重组发生后整个质粒载体整合进入目标基因内部,使目标基因失活。LEENHOUTS等[8-9]称之为Campbell型整合,在乳酸乳球菌中可通过Campbell型质粒进行基因敲除。在对瑞士乳杆菌和戊糖乳杆菌的相关研究中也有单交换失活基因的报道[10-11]。
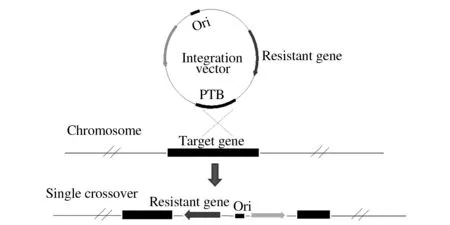
PTB-部分目标基因图1 单交换敲除策略示意图[12]Fig.1 Gene knockout by homologous single crossover
同源重组双交换就是发生两次同源重组,在单交换的基础上再进行一次交换,它分为缺失型突变和插入型突变。通过缺失型突变可以做到无痕敲除,而插入型突变是将目标基因内部插入新的基因序列,使基因失活。除了得到突变型菌株,同源双交换后,还会产生回复突变得到野生型菌株,如图2和图3所示。
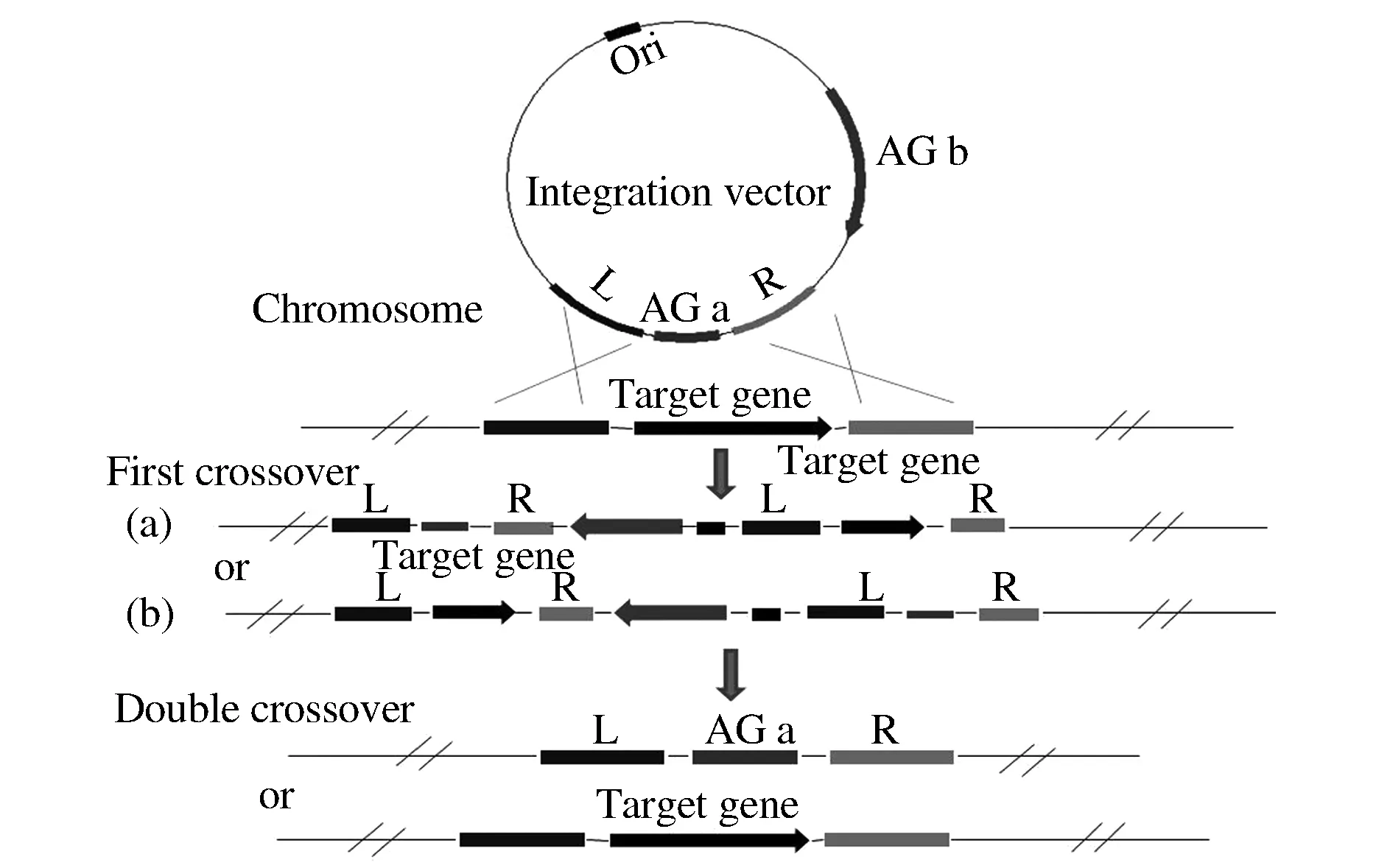
AG-抗性基因;L-左同源臂;R-右同源臂图2 双交换插入型敲除策略示意图Fig.2 Inserted knockout by homologous double crossover
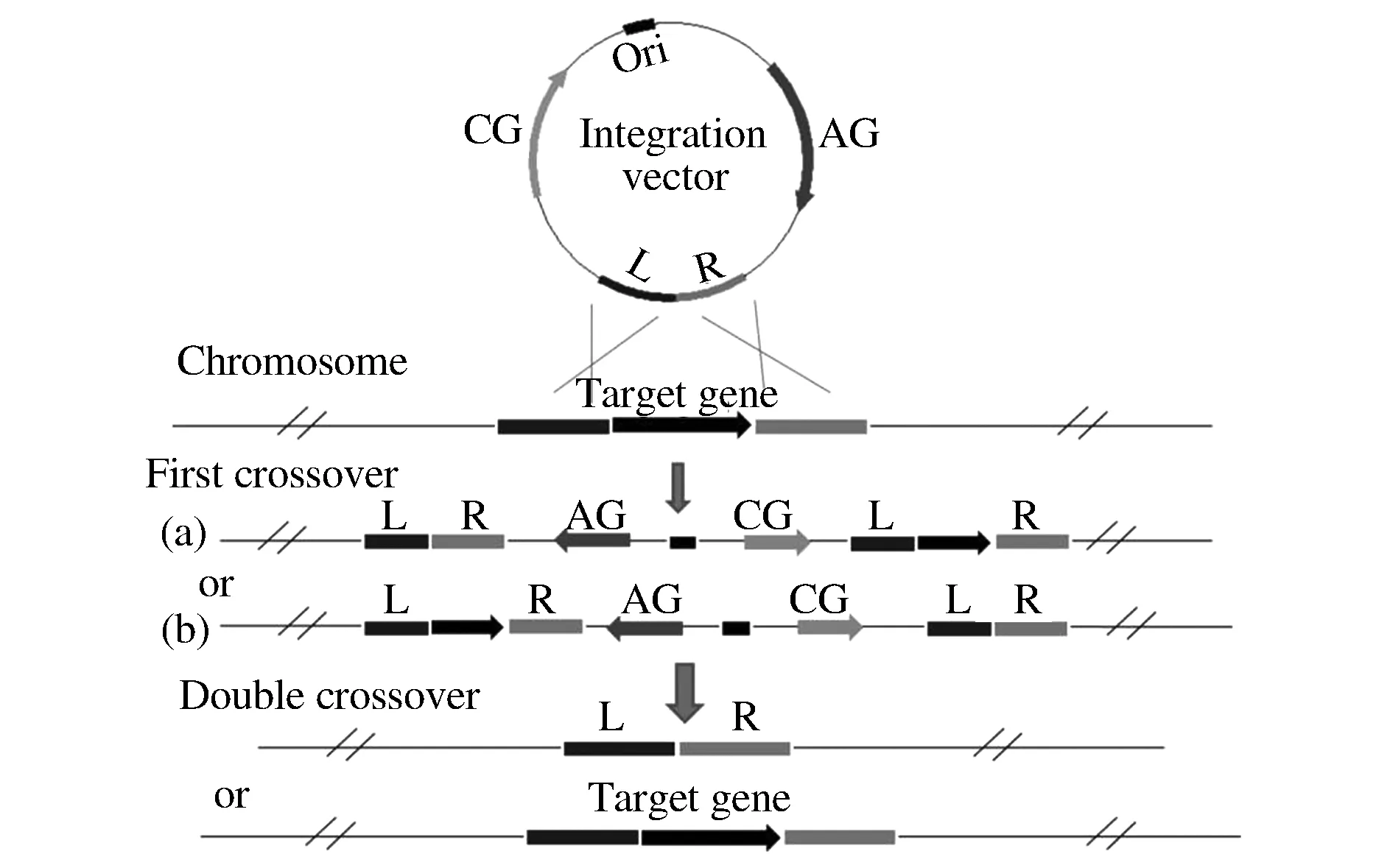
CG-反向筛选标记图3 双交换缺失型敲除策略示意图Fig.3 Markerless knockout by homologous double crossover
早在1996年FERAIN等[13]就运用这种经典的基因敲除方法,利用不能在乳酸菌中复制的pJDC9质粒构建重组载体,对植物乳杆菌染色体上的乳酸脱氢酶L-Idh和D-Idh进行敲除,研究了这两个基因对细胞壁合成的影响。在FERAIN的研究基础上,LIU[14]在2006年对植物乳杆菌TF103再次进行了敲除,利用载体pBluescript构建得到敲除载体,通过同源双交换成功敲除基因als,阻断了丙酮酸到乙酰乳酸的代谢通路,促进了乙醇和甘露醇的生成。2014年JIN等[15]构建了隔离不稳定的敲除载体pCBM32-DSUDs,转化进柠檬明串珠菌中,两次同源重组后,成功把氯霉素基因插入右旋糖酐蔗糖酶基因(dsrC)中。失活dsrC基因的柠檬明串珠菌丧失了合成右旋糖酐的功能,不再具有粘性缺点。研究同时证明了dsrC在蔗糖分解代谢中的重要作用。
同源重组单双交换技术是经典的基因敲除方法,具有很长的应用历史。理论成熟、操作简单,至今仍有较多的学者在研究乳酸菌同源整合载体的构建及鉴定[16-17]。但是这种方法工作量大,敲除效率不高,容易产生回复突变,在革兰氏阳性菌中应用此技术进行基因敲除效率较低。
2.2位点特异性重组敲除
近年来,利用位点特异性重组(site-specific recombination)的基因打靶技术得到了广泛的应用[18]。位点特异性重组系统由重组酶和特异性识别位点两部分构成,重组酶只能识别催化特异性位点间的重组,所以该反应具有高度的专一性和保守性。目前常用于乳酸菌中的位点特异性系统是Cre/loxP系统和TP901-1/att重组系统。
2.2.1Cre/loxP系统
Cre重组酶是位点特异性重组酶家族中的一员,分子质量为38 kDa,能特异性识别DNA序列上的loxP位点。该位点有34个碱基组成,其中包括两个13 bp的反向重复顺序和8bp的间隔区域。Cre/loxP系统主要应用于基因失活或缺失、基因激活、基因易位等,除此之外还可以用于载体的构建。
菌株遗传稳定性是进行遗传操作时需考虑的一个关键因素。基因敲除策略中常引入选择标记基因,从而利于筛选正确的突变体。但如果残留标记基因,可能导致菌体生长迟缓,而且标记基因的表达可能对上下游基因的表达产生影响。因此,在乳酸菌中常用Cre/loxP系统消除基因组上的标记基因。
Cre/loxP系统介导的重组发生后,会在基因组中残留一个loxP位点,当再利用该系统时,会产生不必要的突变。因此,后来发展了一种可以产生稳定迭代重组的突变loxP系统——lox66×lox71Cre系统。当突变的lox66和lox71位点发生重组时,会产生一个退化的lox72位点,该位点不能被Cre酶识别。2007年Michiel等[19]在传统敲除的基础上引入Cre/loxP系统,并使用突变的lox66×lox71位点,成功实现了植物乳杆菌多基因敲除以及筛选标记消除。2014年在Michiel工作的基础上,QIAO等[20]把非复制型质粒pNZ5319转化进入乳酸乳球菌进行同源重组双交换,然后引入Cre/loxP系统消除筛选标记氯霉素,成功敲除乳酸乳球菌NZ9000胸腺嘧啶合成酶基因thyA,获得了食品级筛选标记菌株。所以用Cre/loxP系统从基因组上消除标记基因是一种简洁、有效的方法。
2.2.2TP901-1/att重组酶系统
TP901-1/att重组酶系统是一种能产生稳定单拷贝进行染色体整合的位点特异性重组系统。在此系统中,产生的TP901-1整合酶,会促使两个特异性结合位点高频率重组。这两个不一样的结合位点attB(43bp)和attP(56bp),分别存在于乳酸乳球菌染色体上和构建的整合质粒上[21]。当整合载体和基因组整合完成后,载体的两侧会产生两个杂合的位点attL和attR,如图4(B)所示。尽管该系统具有高效性,但仍存在一些缺点,例如作为筛选标记的抗生素抗性基因残留在染色体上,迭代整合无法完成等。根据attB和attP位置、排列方向的不同,重组后还会产生敲除和倒位的结果。类似于Cre/loxP系统在乳酸菌中的应用,TP901-1/att重组酶系统能消除基因组上的标记基因。结合单交换敲除原理,利用同向的attB和attP,共同敲除目标基因,如图4(A)所示。
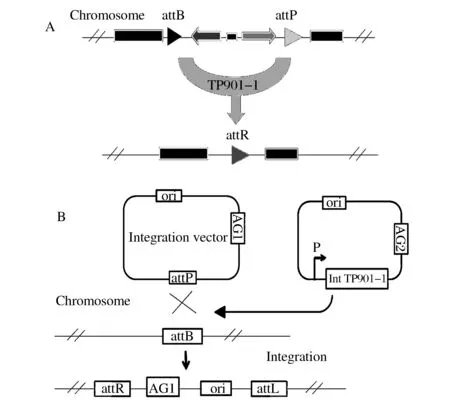
图A-基因敲除;图B-基因整合图4 TP901-1/att重组系统基本原理Fig.4 The site-specific recombination about TP901-1/att
2013年Petersen等[22]构建了一种可以重复迭代整合,无标记的特异位点整合技术。通过构建含有噬菌体结合位点attP的非复制型整合载体pKV6,实现位点特异性打靶。该载体上含有一个最小的细菌结合位点attBmin、一个负筛选标记oroP以及两个用来去除抗性筛选标记的突变lox66×lox71位点。当转入的噬菌体TP901-1整合酶表达时,pKV6会高效的整合进入乳酸菌染色体,通过表达Cre酶,和负筛选标签筛选得到loxP重组体。而引入的最小结合位点attBmin,能够使后续的第二轮的整合成功进行。利用这个工具系统,Petersen等成功把木糖相关基因xylABR和xylT整合进入乳酸乳球菌KF147中,使其能够利用木糖作为碳源。
2.3线形转化敲除法
线形转化敲除是把线性DNA转化进宿主细胞中进行同源重组,是近年来基因敲除方法的热点之一。最为人所熟知的是Red/ET重组系统,常应用于大肠杆菌中。它主要利用λ噬菌体Red蛋白组合(Redα/Redβ/Redγ)和Rec噬菌体诱导表达的蛋白RecE和RecT[23]。与同源双交换相比,该方法所需的同源臂仅为35~50 bp、重组效率更高。但是它主要用于革兰氏阴性菌中,鲜有报道把这个系统应用于乳酸菌中。
目前,能运用于乳酸菌中的线形敲除技术是单链重组工程(Single strand DNA Recombination-SSDR)。它通过诱导表达Red/ET重组系统中的Redβ或者RecT重组酶,把单链核苷酸整合到细菌染色体中,如图5所示。

深灰色的点-抗性基因;灰色椭圆-菌体细胞;R-重组酶基因;EP:表达RecT或Redβ的质粒;Ss DNA-单链DNA图5 单链重组工程示意图Fig.5 Single strand DNA Recombination
Redβ和RecT表达单链DNA结合蛋白,常被作为重组酶,能促进互补DNA链的退火。2012年van Pijkeren等[24-25]在乳酸菌线性重组方面的研究有了突破性的进展,他们在乳酸乳球菌和罗氏乳杆菌中分别构建质粒pJP005和pJP042诱导表达recT基因,转化单链DNA进入细胞,成功突变基因ddl中的几个碱基,使D-丙氨酰-D-丙氨酸连接酶中的一个氨基酸发生改变,从而增强菌株对万古霉素的敏感性。通过MAMA-PCR[26]验证得到突变体,做到了精确定点突变,此方法不需要抗生素及类似筛选标记。
相对于经典的同源双交换敲除,单链重组有更多的优势。它操作简单,重组效率高,不受酶切位点限制,所需同源臂较短,50 bp左右即可,可以直接采用PCR 方法扩增获得。但是这种方法中的重组酶recT在不同菌株中的表达效果不同,不是所有的乳酸菌都适用于这种方法。
3 影响敲除效率的因素
乳酸菌作为革兰氏阳性菌,含有一层较厚的细胞壁,对其进行遗传操作比较困难,所以其重组效率一般较低。但是可以通过优化相关因素提高其敲除效率,例如利用温敏复制子、引入反向筛选标记等。
3.1温敏敲除系统
相对于利用普通非复制型载体进行基因整合,利用温敏敲除系统具有更高的效率。在温敏敲除系统中,敲除载体的复制子为温敏型复制子,转化进入宿主细胞后,在低温下可以让载体复制,高于特定温度时载体丢失,达到消除载体的目的。利用温敏敲除系统,载体在低温复制时,会得到大量拷贝,从而提高二次重组效率。1992年MAGUIN等[27]对广谱型复制子pWV01突变得到了一种突变质粒pVE6002,并把这个温敏复制子应用于敲除载体的构建,发现其具有较高的敲除效率。Biswas等[28]在1993年构建了大肠杆菌-乳酸菌穿梭敲除质粒pGhost5,它在37 ℃时不能复制, 但在28 ℃时能滚环复制。研究表明用这个载体,发生双交换时的剪切效率提高了100~1000倍。这两个载体的发现,极大促进了乳酸菌基因敲除的进展,并沿用至今。最近,Steele等[29]利用pGhost5温敏敲除系统成功敲除掉乳酸乳球菌LM0230的ilvE基因,证明了它在氨基酸代谢中的重要作用。2015年,LANGA等[30]则利用pWV01衍生温敏质粒pVE6007(repA+)和敲除载体pORI28(repA-),共同敲除短乳杆菌pdh30基因,以研究pdh30在甘油代谢中的作用。通过高效液相色谱分析pdh30突变菌株的发酵产物,发现基因pdh30参与3-羟基丙酸的生物合成。
温敏型质粒是温敏敲除系统中很有效的遗传工具,而且其宿主谱较广,适用于许多革兰氏阳性菌。应用温敏敲除系统,先在复制温度下培养,促进转化子的生长繁殖,提高重组效率,重组完成后,改变温度使质粒丢失,通过抗性筛选,得到突变株。但是这个系统遗传学不稳定,而且一般需要高温条件筛选,限制了大部分乳酸菌的生长。
3.2反向筛选标记
运用同源双交换的方法敲除基因,发生第二次重组的效率较低,后期筛选困难。针对该问题,学者们研究发现,采用合适的负筛选标记利于筛选到正确的突变菌株。反向筛选标记能使阳性克隆对一些特殊培养条件敏感,从而促进载体序列丢失和同源重组的发生,提高了筛选效率。同时,利用反向筛选标记还可避免引入抗性基因,达到无痕敲除的目的。
2008年SOLEM等[31]发现了一个可用于乳酸菌的反向标记基因oroP,它编码一种专属的乳清酸运输蛋白(transporter)。oroP基因的存在,会使细胞对5-氟乳清酸敏感,所以它可以作为质粒丢失时的筛选标记。Solem构建了一个含有反向筛选标记基因oroP的载体pCS1966,对乳酸乳球菌IL-1403进行改造,成功把磷酸丙糖异构酶基因(tpiA)前的启动子换成不同的人工合成启动子。他们的研究表明载体pCS1966是种高效的遗传操作工具,而且反向筛选标记基因oroP可广泛应用于乳酸菌、大肠杆菌、枯草芽孢杆菌等多种微生物中。
另一种常运用于乳酸菌中的反向筛选标记基因是upp基因,它广泛存在于原核生物和某些低等真核生物细胞中,表达产物为尿嘧啶磷酸核糖转移酶(UPRT)[32]。UPRT能把5-氟尿嘧啶转化成5-氟单磷酸脱氧尿嘧啶,从而使胸腺嘧啶核苷酸合成酶丧失活性,导致细胞死亡。利用这个特性,upp基因可用作反向筛选标记。早期的研究表明删除upp基因不会影响乳酸菌的嘧啶代谢和生物学特性[33]。2014年Song等[34]应用以温敏复制子pWV01和反向筛选标记upp构建的敲除载体,成功得到干酪乳杆菌ATCC393和乳酸乳球菌MG1363upp基因突变菌株,并发现除了有5-氟尿嘧啶抗性,它在各方面与野生菌株无明显不同。2014年Zhang等[35]人利用温敏质粒pKSV7,先构建upp缺失菌株LL3 Δupp,然后用upp做反向筛选标记,无痕敲除解淀粉芽孢杆菌的基因amyA基因一个47 kb大小的基因簇bae和rocR基因,在很大程度上提高了聚谷氨酸的产量。
upp作为负筛选标记应用广泛,但仍有一些缺陷,许多微生物基因组中都含有upp基因,因此在使用前需改造得到upp突变菌株,然而在某些菌中,upp有可能参与重要代谢,在缺失upp后,有可能会影响菌株生长。
3.3电转化方法优化
通过优化电转化方法,提高转化效率,使得重组载体更容易转化进入乳酸菌细胞,随之可提高重组的效率。因为乳酸菌是革兰氏阳性菌,被一层致密的厚细胞壁包裹着,所以转化乳酸菌细胞时一般使用电转化法和原生质体转化法。原生质体转化法需要先去细胞壁,操作繁杂且转化效率低,所以转化乳酸菌更常用省时简便的电转化法。影响电转化效率的因素较多,如电击的电压、乳酸菌所处的生长周期、电击缓冲液等。
Gerber等[36]先优化了电转乳酸乳球菌IL1403的具体步骤,最后使转化效率达到106CFU/μg DNA,然后再构建以pCR-Blunt IITopo载体衍生的pSG3和pSG7重组质粒,把它们电转化进入乳酸乳球菌后,重组效率是每微克质粒DNA产生大约10个阳性克隆,成功敲除掉基因copA和copB。2012年Spath等[37]发现利用非甲基化的质粒电转化植物乳杆菌时,转化效率得到极大的提高,在此基础上进行的同源重组,效率提高明显。
由于不同种乳酸菌菌株的差异性,具体的电转化优化条件需要在实验过程中去摸索,比如两株不同的乳酸菌最适电压可能存在很大差异。当电转化达到一个理想的效果,再进行基因敲除,也会使敲除效率得到提高。
3.4其他因素
利用重组载体打靶时,同源臂的长度是影响重组效率的一项重要因素。LELOUP等[38]发现在沙克乳杆菌中,同源臂长度1 kb以内,重组效率随同源臂长度增加呈线性增长,大于1 kb后,再增加同源臂长度,重组效率无明显增高。同样GERBER等[36]也在乳酸乳球菌IL1403中做了相关研究,得到相同的结果。
另外,利用单链DNA进行重组时,也存在一些因素影响重组效率,例如单链DNA的浓度、单链的类型、修饰末端等。vanPijkeren等[39]通过优化,发现在乳酸乳球菌中,使用滞后链的重组效率比用前导链高25倍,提高寡核苷酸的浓度到500 μg并用硫代磷酸修饰末端后,重组效率提高了10倍。
4 结论与展望
大部分乳酸菌以益生特性著称,作为一种重要的工程菌应用于医药食品行业[40]。在分子生物学中,基因敲除技术是生物育种的重要技术支持,也是对乳酸菌进行遗传改造的重要方法。目前常用于乳酸菌的敲除策略是传统的同源重组双交换敲除,但它具有重组效率低、耗时较长、工作繁杂等缺点。随着现代技术手段的进步,学者们通过优化重组载体和转化效率,在提高敲除效率方面取得了进步。另一方面,在乳酸菌中对单链重组工程方面的研究也取得了不错的进展,可以更加高效便捷的敲除基因,尽管在适用性方面有一定的限制,但这些研究都为未来乳酸菌基因敲除技术的发展奠定了基础。
随着分子生物学的发展,近年来新兴的基因敲除技术成为研究热点。其中,CRISPR/Cas技术[41]未来极有可能应用于乳酸菌中。应用该系统WANG 等[42]已经成功无痕敲除同样是革兰氏阳性菌的贝氏梭状芽胞杆菌基因spo0A。van Pijkeren等[43]已尝试把CRISPR/Cas系统和单链重组系统结合起来应用于乳酸菌中。MILLEN[44]等的研究从遗传水平上表明CRISPR/Cas系统在乳酸菌细胞内的有效性,及广阔的应用前景。除此之外,RNA干扰(RNAi)[45]、TALENS[46](Transcription Activator-like (TAL) Effector Nucleases)、锌指核酸酶基因打靶技术[47](Zinefinger-nucleases,ZFNs)等高效靶向基因修饰技术都有待应用于乳酸菌中,这将极大的提高基因敲除效率,使乳酸菌的遗传操作更加便捷。
[1]PETERBAUER C,MAISCHBERGER T,Haltrich D.Food-grade gene expression in lactic acid bacteria[J]. Biotechnology J ournal,2011,6(9):1 147-1 161.
[2]DELAYE L,GONZLEZ-DOMENECH C M,GARCILLN-BARCIA M P,et al.Blueprint for a minimal photoautotrophic cell: conserved and variable genes inSynechococcuselongatusPCC 7942[J]. BMC Genomics,2011,12(1): 25.
[3]HASHIMOTO M,ICHIMURA T,MIZOGUCHI H,et al.Cell size and nucleoid organization of engineeredEscherichiacolicells with a reduced genome[J].Molecular Microbiology,2005,55(1):137-149.
[4]MORIMOTO T,KADOYA R,ENDO K,et al.Enhanced recombinant protein productivity by genome reduction inBacillussubtilis[J].DNA Research,2008,15(2):73-81.
[5]KOMATSU M,UCHIYAMA T,MURA S,et al.Genome-minimizedStreptomyceshost for the heterologous expression of secondary metabolism[J].Proceedings of the National Academy of Sciences,2010,107(6):2 646-2 651.
[6]焦晶凯,刘振民,莫蓓红.乳制品乳酸菌遗传学概述[J].食品与发酵工业,2014,40(1): 160-167.
[7]张霖,赵国屏,丁晓明.位点特异性重组系统的机理和应用[J].中国科学:生命科学,2011(12): 1 090-1 111.
[8]LEENHOUTS K J,KOK J,VENEMA G.Campbell-like integration of heterologous plasmid DNA into the chromosome ofLactococcuslactissubsp. lactis[J].Applied and environmental Microbiology, 1989,55(2):394-400.
[9]LEENHOUTS K J,KOK J,VENEMA G. Replacement recombination inLactococcuslactis[J].Journal of Bacteriology, 1991, 173(15):4 794-4 798.
[10]BHOWMIK T,STEELE J L.Development of an electroporation procedure for gene disruption inLactobacillushelveticusCNRZ 32[J].Journal of General Microbiology,1993,139(7):1 433-1 439.
[11]LOKMAN B C,HEERIKHUISEN M,LEER R J,et al.Regulation of expression of theLactobacilluspentosusxylAB operon[J].Journal of Bacteriology,1997,179(17):5 391-5 397.
[12]于慧敏,马玉超.工业微生物代谢途径调控的基因敲除策略[J].生物工程学报,2010,26(9): 1 199-1 208.
[13] FERAIN T,HOBBS J N,RICHARDSON J,et al.Knockout of the twoldhgenes has a major impact on peptidoglycan precursor synthesis inLactobacillusplantarum[J].Journal of Bacteriology,1996, 178(18):5 431-5 437.
[14]LIU S.A simple method to generate chromosomal mutations inLactobacillusplantarumstrain TF103 to eliminate undesired fermentation products[J].Applied Biochemistry and Biotechnology, 2006,131(1-3):854-863.
[15]JIN Q,LI L,KIM Y J,et al.Construction of a dextran-freeLeuconostoccitreummutant by targeted disruption of the dextransucrasegene[J].Journal of Applied Microbiology,2014,117(4): 1 104-1 112.
[16]秦艳青,金宁一,李昌,等.乳酸乳球菌同源整合载体的构建及鉴定[J].吉林农业大学学报, 2012,34(6):628-633.
[17]李娅妮,赵国芬,包秋华,等.植物乳杆菌lai基因同源重组敲除载体的构建与鉴定[J].农产品加工: 学刊 (下),2014 (3):51-53.
[18]GRINDLEY N D F,WHITESON K L,RICE P A.Mechanisms of site-specific recombination[J].Annu Rev Biochem,2006,75:567-605.
[19]LAMBERT J M,BONGERS R S,KLEEREBEZEM M. Cre-lox-based system for multiple gene deletions and selectable-marker removal inLactobacillusplantarum[J].Applied and Environmental Microbiology, 2007,73(4):1 126-1 135.
[20]ZHU D,ZHAO K,XU H,et al.Construction ofthyA deficientLactococcuslactisusing the Cre-loxPrecombination system[J].Annals of Microbiology,2015,65(3):1 659-1 665.
[21]BRØNDSTED L,HAMMER K.Use of the integration elements encoded by the temperate lactococcal bacteriophage TP901-1 to obtain chromosomal single-copy transcriptional fusions inLactococcuslactis[J]. Applied and Environmental Microbiology,1999,65(2):752-758.
[22]PETERSEN K V,MARTINUSSEN J,JENSEN P R,et al.Repetitive, marker-free, site-specific integration as a novel tool for multiple chromosomal integration of DNA[J].Applied and Environmental Microbiology,2013,79(12):3 563-3 569.
[23]王军平,张友明.Red/ET 重组及其在生物医学中的应用[J].生物工程学报,2005,21(3): 502-506.
[24]van PIJKEREN J P,BRITTON R A.High efficiency recombineering in lactic acid bacteria[J]. Nucleic Acids Research,2012,40(10):e76-e76.
[25]van PIJKEREN J P,BRITTON R. Precision genome engineering in lactic acid bacteria[J].Microbial Cell Factories,2014,13(Suppl 1):S10.
[26]QIANG Y Z,QIN T,FU W,et al. Use of a rapid mismatch PCR method to detectgyr aandpar cmutations in ciprofloxacin-resistant clinical isolates ofEscherichiacoli[J].Journal of Antimicrobial Chemotherapy,2002,49(3):549-552.
[27]MAGUIN E,DUWAT P,HEGE T,et al.New thermosensitive plasmid for gram-positive bacteria[J]. Journal of Bacteriology,1992,174(17):5 633-5 638.
[28]BISWAS I,GRUSS A,EHRLICH S D,et al.High-efficiency gene inactivation and replacement system for gram-positive bacteria[J].Journal of Bacteriology, 1993,175(11):3 628-3 635.
[29]ATILES M W,DUDLEY E G,STEELE J L.Gene cloning, sequencing, and inactivation of the branched-chain aminotransferase ofLactococcuslactisLM0230[J].Applied and Environmental Microbiology,2000,66(6):2 325-2 329.
[30]LANGA S,ARQUÉS J L,GAYA P,et al.Glycerol and cobalamin metabolism in Lactobacilli: relevance of the propanediol dehydrogenasepdh30[J].European Food Research and Technology, 2015,241(2):173-184.
[31]SOLEM C,DEFOOR E,JENSEN P R,et al.Plasmid pCS1966, a new selection/counterselection tool for lactic acid bacterium strain construction based on theoroPgene, encoding an orotate transporter fromLactococcuslactis[J].Applied and Environmental Microbiology,2008,74(15): 4 772-4 775.
[32]MARTINUSSEN J,HAMMER K.Cloning and characterization ofupp, a gene encoding uracil phosphoribosyltransferase fromLactococcuslactis[J].Journal of Bacteriology,1994,176(21): 6 457-6 463.
[33]KRISTICH C J,MANIAS D A,DUNNY G M. Development of a method for markerless genetic exchange inEnterococcusfaecalisandits use in construction ofasrtAmutant[J].Applied and Environmental Microbiology,2005,71(10):5 837-5 849.
[34]SONG L, CUI H, TANG L, et al.Construction of upp deletion mutant strains ofLactobacilluscaseiandLactococcuslactisbased on counterselective system using temperature-sensitive plasmid[J].Journal of Microbiological Methods,2014,102: 37-44.
[35]ZHANG W,GAO W,FENG J,et al.A markerless gene replacement method forB.amyloliquefaciensLL3 and its use in genome reduction and improvement of poly-γ-glutamic acid production[J]. Applied Microbiology and Biotechnology,2014,98(21):8 963-8 973.
[36]GERBER S D,SOLIOZ M.Efficient transformation ofLactococcuslactisIL1403 and generation of knock-out mutants by homologous recombination[J].Journal of Basic Microbiology,2007,47(3): 281-286.
[37]SPATH K,HEINL S,GRABHERR R.Direct cloning inLactobacillusplantarum: Electroporation with non-methylated plasmid DNA enhances transformation efficiency and makes shuttle vectors obsolete[J].Microb Cell Fact,2012,11:141.
[38]LELOUP L,EHRLICH S D,ZAGOREC M,et al.Single-crossover integration in theLactobacillussakechromosome and insertional inactivation of theptsI andlacLgenes[J].Applied and Environmental Microbiology,1997, 63(6):2 117-2 123.
[39]van PIJKEREN J P,NEOH K M,SIRIAS D,et al.Exploring optimization parameters to increase ssDNA recombineering inLactococcuslactisandLactobacillusreuteri[J].Bioengineered,2012,3(4): 209-217.
[40]郭志华,杨洪.分离自藏灵菇的乳酸菌的益生特性[J].食品与发酵工业,2013,39(1):151-154.
[41]BARRANGOU R,FREMAUX C,DEVEAU H,et al.CRISPR provides acquired resistance against viruses in prokaryotes[J].Science,2007,315(5 819):1 709-1 712.
[42]WANG Y,ZHANG Z T,SEO S O,et al.Markerless chromosomal gene deletion inClostridiumbeijerinckiiusing CRISPR/Cas9 system[J].Journal of Biotechnology,2015,200:1-5.
[43]OH J H,van PIJKEREN J P.CRISPR-Cas9-assisted recombineering inLactobacillusreuteri[J]. Nucleic Acids Research,2014:doi:10.1093/nar/gku623.
[44]MILLEN A M,HORVATH P,BOYAVAL P,et al.Mobile CRISPR/Cas-mediated bacteriophage resistance inLactococcuslactis[J].PLoS One,2012,7(12):e51663.
[45]JACKSON A L,BARTZ S R,SCHELTER J,et al.Expression profiling reveals off-target gene regulation by RNAi[J].Nature Biotechnology,2003,21(6):635-637.
[46]CERMAK T,DOYLE E L,CHRISTIAN M,et al.Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting[J].Nucleic Acids Research,2011:doi:10.1093/nar/gkr 218.
[47]MILLER J C,HOLMES M C,WANG J,et al.An improved zinc-finger nuclease architecture for highly specific genome editing[J].Nature Biotechnology,2007,25(7):778-785.
Advances on gene knockout technology of lactic acid bacteria
DU Sheng-yang,WANG Bin-bin,FENG Jia,HOU Bin-xiao,QIAO Jian-jun*
(Department of Pharmaceutical Engineering, School of Chemical Engineering and Technology, Key Laboratory of Systems Bioengineering, Ministry of Education, Tianjin University, Tianjin 300072,China)
Lactic acid bacteria(LAB)are Gram-positive bacteria that have been widely used in food, medicine, light industry, feed industry and many other industries. The genetic manipulation of LAB is the foundation of gene regulation and metabolism. We can identify the function of the unknown genes in the genome and assure the application of its encoding product through the gene knockout technology. Besides, the heterologous gene products of LAB can be obtained more purposefully and selectively. Currently, gene knockout has become a newly-developed molecular biological technology in LAB. This article mainly summarized the advances of gene knockout technology of LAB and its efficiencies. The existing problems were analyzed and the development trend of gene knockout of LAB was preliminary discussed.
gene knockout; lactic acid bacteria; homologous recombination; single-strand DNA recombineering; knockout efficiency
10.13995/j.cnki.11-1802/ts.201601044
硕士研究生(乔建军为通讯作者,E-mail:jianjunq@tju.edu.cn)。
国家自然科学基金项目(31270142);国家科技支撑计划(2014BAD02B00)
2015-05-18,改回日期:2015-06-20
