超高效液相色谱串联质谱法对水产品中硝基呋喃代谢物的测定
2016-09-24王狄张嘉楠
王狄 张嘉楠
摘要:建立了使用Waters UPLC Xevo TQ超高效液相色谱串联质谱仪检测水产品中4种硝基呋喃代谢物的分析方法。匀浆后的样品在酸性环境下水解,并用2一硝基苯甲醛衍生16h,经磷酸氢二钾提取,LMS固相萃取小柱净化后,在三重四极杆多反应监测模式下进行测定。方法采用同位素内标法定量,四种代谢物的检出限(LOD)均低于0.25μg·kg-1,线性范围在0.5μg·L-1到10μg·L-1间,相关系数大干0.99。在0.5、1.0和5μg·kg-1浓度下分别进行添加回收试验(n=5),回收率在75.2%~123.3%之间,相对标准偏差(RSD)小于9.4%。
关键词:硝基呋喃代谢物;固相萃取;超高效液相色谱串联质谱法;水产品
硝基呋喃类药物(nitrofurans)主要是指呋喃唑酮(furazolidone,AOZ)、呋喃它酮(furMta-done,AMOZ)、呋喃西林(nitrofurazone,SEM)、呋喃妥因(nitrofurantoin,AHD)等引入硝基的人工合成抗菌药,近年来发现其具有慢性毒性,可引起消化道反应,美国、欧盟等工业发达国家和地区已禁止使用。硝基呋喃类药物在动物体内数小时内可降解,但其代谢产物能与蛋白质紧密结合,形成稳定的残留物,残留时间达数周之久,甚至在蒸煮、烘烤、磨碎和微波加热过程中也无法有效降解,因此检测硝基呋喃类药物的代谢产物,更能起到监控作用。目前欧盟对硝基呋喃类代谢物的检测方法灵敏度规定为1μg/kg。
试验采用了超高效液相色谱一串联质谱法检测硝基呋喃类药物代谢物。在农业部781号公告-4-2006的基础上对样品前处理过程进行了简化,并且采用固相萃取法对样品进行净化,提高了工作效率和实验结果的稳定性。经过多次试验证明方法的结果及稳定性可靠,适用于日常水产品中硝基呋喃代谢物的痕量检测工作。
1 材料与方法
1.1 仪器与试剂
UPLC Xevo TQ液相色谱串联质谱仪(美国沃特世);双功能气浴恒温振荡器(江苏天由有限公司);台式高速冷冻离心机(湖南赫西仪器装备有限公司);氮空气吹扫浓缩仪(北京斯珀特科技有限公司);CGX一274ASPEC四通道自动固相萃取仪(法国吉尔森)。
甲醇、乙腈、甲酸和正己烷均为色谱纯;盐酸为优级纯;磷酸氢二钾为分析纯;2-硝基苯甲醛(购自德国CNW公司);所有标准品及同位素内标均购自德国Dr.Ehrenstorfer;Bond Elut LMS固相萃取小柱(200mg,3mL)。
1.2 样品前处理
衍生化:称取(2±0.05)g样品于离心管中,加入100μg·L-1的混合内标标准溶液100μL,再加入0.1mol·L-1邻硝基苯甲醛100μL、0.2mol·L-1的盐酸5mL,漩涡混合30s后,于37℃恒温箱中避光衍生16h。
提取:待样品冷却至室温后,加入1mol·L-1磷酸氢二钾5mL,漩涡混匀,用1mol·L-1氢氧化钠调pH值至7.2~7.4后,8000r/min离心10min。
净化:将5mL样品上清液加入到BondElutLMS固相萃取小柱中(使用前依次用3mL乙酸乙酯、甲醇和水活化),然后用3mL水淋洗、吹干,最后用3mL乙酸乙酯洗脱。洗脱液于40℃氮气流下吹干,残渣用1mL0.1%甲酸水溶解,过0.22μm微孔滤膜后上机测定。
1.3 仪器条件
液相色谱条件:
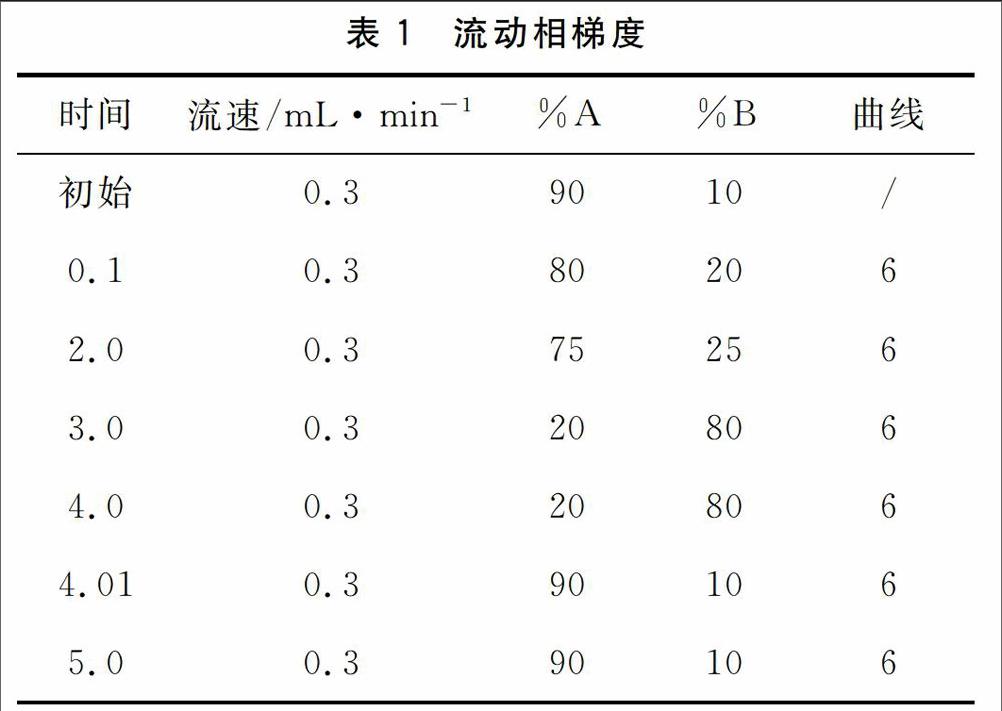
色谱柱:ACQUITY UPLC BEH C18 1.7μm 2.1×100 mm;进样量:5μL;流动相:A:0.1%甲酸水B:乙腈;流速:0.3mL/min;柱温:30℃
质谱条件:
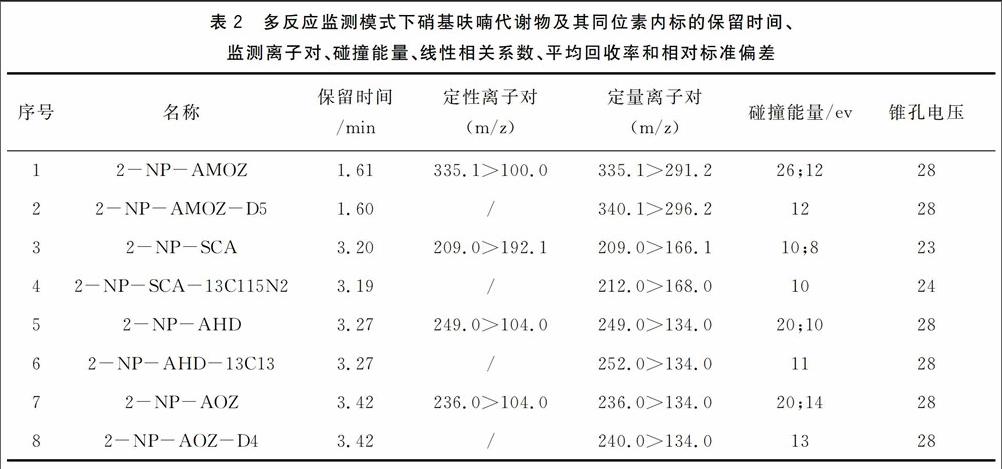
离子源:ESI;检测方式:多反应监测;脱溶剂气流量:650L/h;锥孔气流量:50L/h;脱溶剂气温度:350℃;毛细管电压:3kV;离子源:150℃;碰撞气流量:0.18mL/min;正离子模式;其它参数见表2。
2 结果与讨论
2.1 提取方法的选择
试验在农业部781号公告-4-2006前处理方法的基础上加入了固相萃取净化过程。由于乙酸乙酯较水极性小,所以在固相萃取过程中,极性较小的乙酸乙酯作为溶剂会一定程度上破坏非极性药物和非极性吸附剂之间的非极性作用力。因此,在提取过程中简化掉了药物从水溶液到乙酸乙酯的转移过程,同时降低了药物在转移过程中的损失,提高了回收率。
2.2 固相萃取小柱的选择
BondElut LMS:是聚合物吸附剂,萃取机制为非极性,萃取化合物为非极性化合物,在洗脱样品时无须再添加有机胺类改性剂、缓冲液或者酸等。次级相互作用的消除意味着使用纯的有机溶剂或与HPLC流动相兼容的低离子强度的洗脱液即可洗脱目标分析物。这种特性可以很好地保证萃取技术与LC/MS和其它高灵敏度的检测技术的兼容性。优选的吸附剂颗粒大小和大的比表面积保证了萃取效率,可以实现无干扰萃取。实验参考了Alexander Leitner,Peter Zollner,Wolfgang LindnerE<对样品的固相萃取净化方法,并选择了与其方法所用的固相萃取小柱填料相同的安捷伦Bond Elut LMS固相萃取小柱(200mg,3mL)。通过吉尔森四通道自动固相萃取仪对不同添加水平的样品进行固相萃取测定试验,以及相同添加浓度的样品分别进行固相萃取和无固相萃取过程的对比试验,结果表明,BondElutLMS固相萃取小柱能够有效地去除样品基质中蛋白类基质的干扰,并且能够保证高的回收率和结果重现性。
2.3 方法的线性关系、精密度及准确度
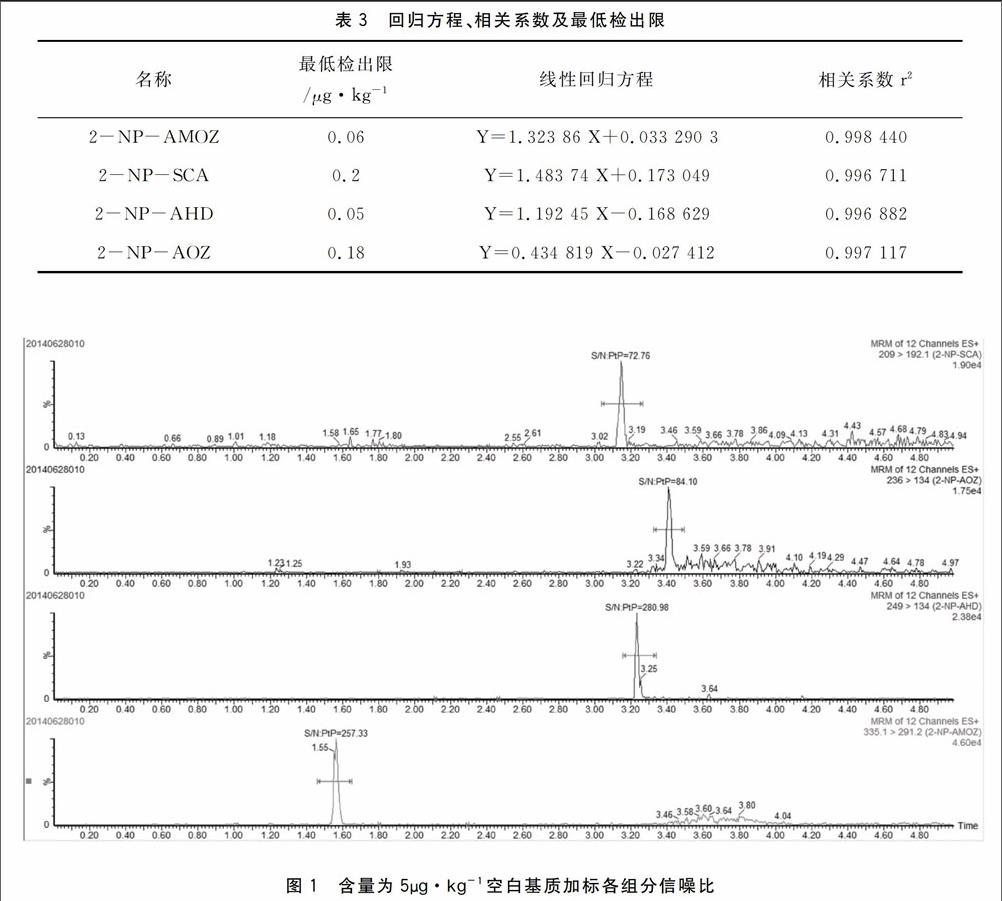
按照实验的条件和方法,配置浓度分别为0.5、1、2、5、10μg·L-1一系列标准溶液进行测定,用定量离子峰面积与内标峰面积比和标准溶液浓度和内标浓度比做校正曲线,线性回归方程、相关系数及最低检出限见表3。通过空白基质加标,并按照S/N=3计算方法检出限。结果表明,4种硝基呋喃代谢物的检出限为0.05-0.2μg·kg-1。
2.4 加标回收及精密度实验
实验选用大菱鲆、草鱼、对虾为基质,分别采用0.5、1.0和5μg·kg-1三个加标水平,计算平均回收率和精密度(n=5),结果见表4。化效果好,能够对水产品中的硝基呋喃代谢物进行准确的定性和定量分析。
(收稿日期:2016-05-16;~回日期:2016-05-23)
