Interactions of Anionic and Neutral Serine with Pure and Metal-doped Graphene Studied by Density Functional Theory
2016-09-23QunWang,Meng-haoWang,Ke-fengWang等
Interactions of Anionic and Neutral Serine with Pure and Metal-doped Graphene Studied by Density Functional Theory
I.INTRODUCTION
Graphene is a two-dimensional atomic layer of carbon atoms in hexagonal arrangement.Recently,graphene has attracted significant attention in the development of sensors owing to its unique structural,chemical,and electrical properties[1,2].Graphene is commonly modified by doping with covalent or non-covalent functionalization to improve the characteristics of graphene-based sensors and electronic devices[3,4].Researches showed that doping graphene with metals such as Au,Ni,Al,Mn,or Cu,can effectively modify the performance of graphene-based sensors.Cazorla[5]investigated interactions of glycine(Gly),proline(Pro),and hydroxyproline(Hyp)with pure and Ca-doped graphene,and found that interactions of these amino acids on the Ca-doped graphene surface are stronger than that on the pure graphene surface.Zhang et al.[6]indicated that the interaction of cysteine(Cys)with a Audoped graphene surface is stronger than that on the unmodified graphene surface,due to the formation of strong Au-O,Au-S,and Au-N interactions.Ma et al.[7]discovered that adsorption of Cys on a Pt-doped graphene surface is greater than that on the pristine graphene surface.Chi et al.[8]studied the adsorption of HCHO on pure and Al-doped graphene surfaces,and suggested that the Al-doped graphene can be more suitable for the detection of HCHO gas.Ⅰn addition,many researchers have investigated interactions between small gas molecules such as CO,H2O,NH3,NO2,and HCHO,and pure or doped graphene.They discovered that the doped graphene can be more sensitive to these gas molecules[8-11].
The structure and stability of Ser have been studied intensively in the last decade using theoretical and experimental methods,because of the biological significance[12,13].Ser plays an important role in the metabolism of lipids,fatty acids,and muscle growth by producing immunoglobulin and antibodies,thus maintaining a healthy immune system.
To the best of our knowledge,previous theoretical studies about the interaction between Ser and graphenebased materials have not been reported.Ⅰn this work,in order to explore potential applications of metal-doped graphene sensors,and to provide a useful reference for designing and developing graphene-based sensors for t-he different forms of Ser,we study systematically interactions of the anionic and neutral Ser with pure and metal-doped graphene(G)by the density functional theory(DFT)method.The stable adsorption configurations,adsorption energies,the density of states (DOSs),the Mulliken atomic charges,and differences of electron density are all discussed in detail.
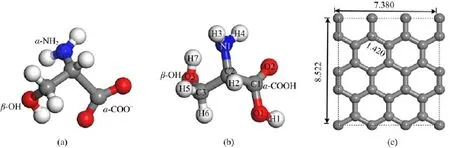
FⅠG.1 Three-dimensional structures of(a)anionic Ser,(b)neutral Ser,and(c)G.Carbon:gray,oxygen:red,nitrogen: blue,hydrogen:white.Bond lengths are in˚A.
II.COMPUTATIONAL DETAILS
A.Building models
SerandGmodels:Thechemicalformulaof anionic and neutral Ser are-OOCCH(NH2)CH2OH and HOOCCH(NH2)CH2OH,respectively.The Ser molecules contain one α-amino group(α-NH2),one α-carboxyl group(α-COO-or α-COOH)and one βhydroxyl group(β-OH).Three-dimensional structure models are shown in Fig.1(a)and(b).The G model contains 16 six-member carbon rings,for 32 carbon atoms in total.The C-C bond length is 1.420˚A,which is consistent with the previous results[14,15].A larger supercell with 96 atoms has also been used to test the calculated results.We found that our results are not affected by the supercell size,which accord with Zhang et al.’s results,they found that interactions of NO2,NO and Ti,N-doped graphene are not affected by the size of graphene[16].Ⅰn fact,the 32 atoms supercell is usually used to simulate graphene[8,15,17,18].Periodic boundary conditions are used to model the slab,and the area of the surface is 8.522˚A×7.380˚A.A vacuum slab 25˚A in thickness is added to separate each slab from its periodic images,as shown in Fig.1(c).
Metal-doped G models:Based on the model of pure G,metal-doped G models are built by replacing a carbon atom with a metal atom.The optimized structural models of metal-doped G are shown in Fig.2.
Pure and metal-doped G-Ser models:the anionic and neutral Ser interact with pure and metal-doped G. When the α-NH2of Ser directed the Fe-doped surfaces,the adsorption energies are quite small(-0.346 eV for the anionic Ser and-0.268 eV for the neutral Ser), therefore,the α-COO-,α-COOH and-OH of Ser adjacent to these G surfaces models are built.
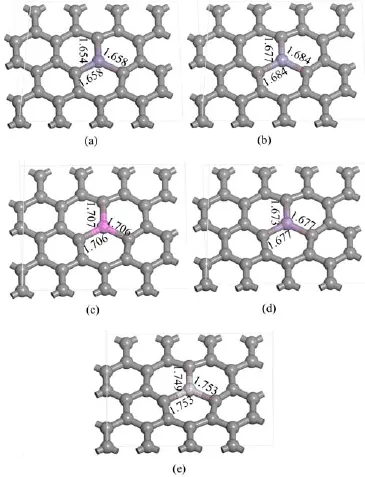
FⅠG.2 Structures of(a)Fe-,(b)Cr-,(c)Al-,(d)Mn-,(e) Ti-doped G after geometric structure optimization.Bond lengths are in˚A.
B.Simulation parameters
The simulation was performed with the DFT program Dmol3in Materials Studio(Accelrys,San Diego,CA),in which the physical wave functions were expanded in terms of numerical basis sets.Dmol3produces highlyaccurate results,while keeping the computational cost fairly low[19,20].The bond lengths and bond angles of neutral Ser from Dmol3(TableⅠ),are in agreement with previous results[21].The reproducibility of previously reported data validates the applicability of Dmol3to the present systems.
During simulation,the DNP double numerical basis set was used,which is comparable to the 6-31G∗∗basis set.Core electrons were treated with DFT semi-core pseudo potentials[22].The exchange-correlation energy was calculated using the Perdew-Burke-Ernzerh of generalized gradient approximation,the GGA of PBE can be defined as:a detailed description of the GGA of PBE can be found in Ref.[23].Special point sampling integration over the Brillouin zone was employed using the Monkhorst-Pack scheme with a 2×2×1 k-point mesh.A Fermi smearing of 0.005 Ha(1 Ha=27.211 eV),and a global orbital cutoff of 4.8˚A were employed.We left thespin unrestricted for the entire systems,and the TS method for DFT-D corrections was used to deal with van der Waals forces in the simulation systems[24]. The convergence criteria for the geometry optimization and energy calculation were set as follows.The selfconsistent field tolerance was 10-6Ha/atom,energy tolerance was 10-5Ha/atom,a maximum force tolerance was 0.002 Ha/˚A,and a maximum displacement tolerance was 0.005˚A.

FⅠG.3 Anionic Ser interacting with the(a)pure G and(b)Fe-,(c)Cr-,(d)Al-,(e)Mn-,(f)Ti-doped G.Bond lengths are in˚A.
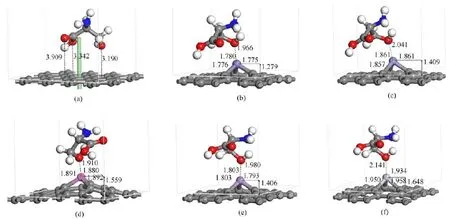
FⅠG.4 Neutral Ser interacting with the(a)pure G and(b)Fe-,(c)Cr-,(d)Al-,(e)Mn-,(f)Ti-doped G.Bond lengths are in˚A.
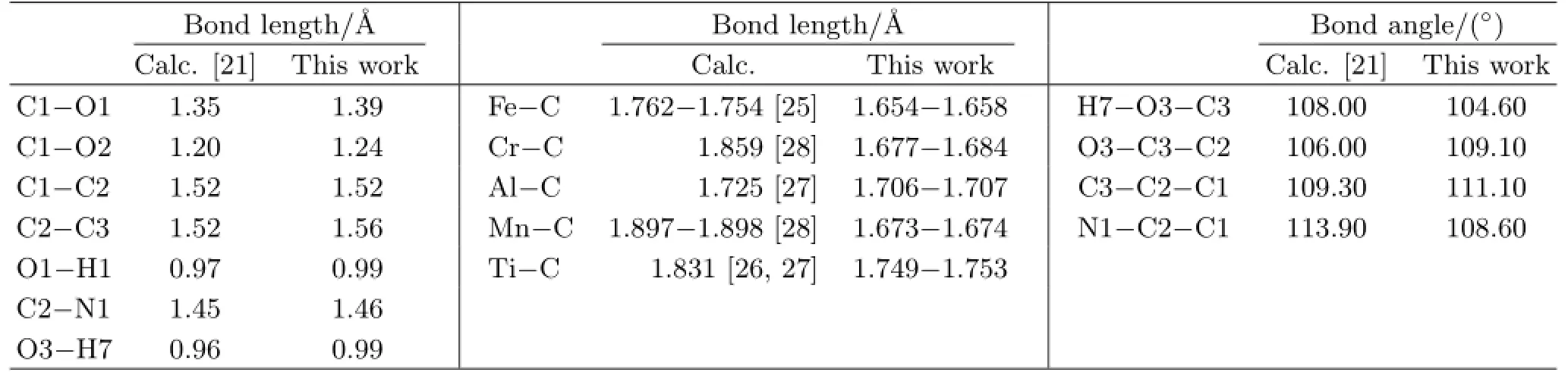
TABLEⅠComparison of results in bond lengths and angles of the neutral serine molecule and bond lengths of metal-C from literature values calculated with Dmol calculations.
C.Adsorption energy
The adsorption energy of Ser is calculated according to the following equation:
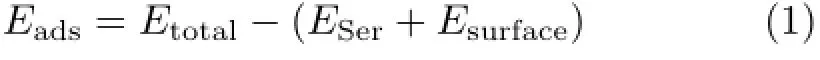
Where Etotal,ESer,and Esurfacerepresent the total energy of the adsorption system,the energy of the Ser molecule,and the energy of the graphene surface,respectively.A negative Eadsvalue corresponds to a stable adsorption.The more negative Eadsis,the more stable the adsorbed structure is.
D.Differences in electron density
Electron density maps can directly display the interaction between biomolecules and biomaterials.The electron density difference reveals the change of electron density in the process of adsorption,which is calculated by the total electron density(ρtotal),subtracting the electron density of isolated Ser(ρSer),and the G surface(ρsurface),
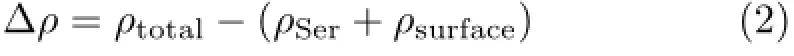
III.RESULTS AND DISCUSSION
A.Stability analysis of the metal-doped graphene models
Metal-doped graphene models are built by replacing a carbon atom with a metal atom,after the structure of graphene is optimized.Then,the metal-doped graphene models are optimized to estimate their stabilities,as shown in Fig.2.The doped atoms did not protrude out of the graphene plane,and the distances between C and the metals increase comparable with the C-C distance.The distances are Fe-C:1.654-1.658˚A,Cr-C:1.677-1.684˚A,Al-C:1.706-1.707˚A,Mn-C: 1.673-1.674˚A,and Ti-C:1.749-1.753˚A,which are in accord with previous studies(TableⅠ)[25-28].Furthermore,these also suggested that the bonds of metal to carbon are not destroyed,and the metal-doped graphene models in this study are all stable.
B.Analysis of adsorption energies
Both anionic and neutral Ser lay parallelly to the pure and metal-doped graphene surfaces and the α-COOH,α-COO-and-OH of Ser are adjacent to the metal-doped G surfaces.Ⅰnteractions of both types of Ser on pure graphene surfaces are weak because of the low adsorption energies of-0.75 and-0.53 eV for anionic and neutral Ser,respectively.Moreover,we found weak non-covalent interactions due to the formation of -COOH···π,-COO-···π,and-OH···π interaction between Ser and pure graphene,as shown in Fig.3(a)and Fig.4(a).Ⅰn the pure G-anionic Ser model,the shortest distance between the O atom of-OH and the graphene is 3.189˚A.The O atoms of-COO-are oriented toward the graphene surface,and the shortest distances between the O atoms and the graphene are 3.184 and 3.081˚A,respectively,as shown in Fig.3(a).Ⅰn the pure G-neutral Ser model,the shortest distance between the O atom of -OH and graphene is 3.190˚A,and the shortest distances between the O atoms of-COOH and graphene are 3.342 and 3.909˚A,respectively,as shown in Fig.4(a).These results are comparable with those previous investigations on the interaction between amino acids or peptides and graphene.Guo et al.found that distances are between the 2.649 and 3.660˚A when the RGD peptide interacts with pure graphene[14].Ma et al.found that the distances between graphene and Cys vary from 3.280˚A to 3.960˚A[7].The distances in this work follow the non-covalent-COO-···π,-COOH···π and-OH···π interaction standard,which shows weak noncovalent interactions between both types of Ser and pure graphene[29,30].
TableⅠⅠshows adsorption energies of all models.The interactions are much stronger between both types of Ser and metal-doped graphene surfaces than that between both types of Ser and pure graphene surfaces. Moreover,they are typical of covalent interactions between the Ser and metal-doped graphene due to the formation of strong metal-O and O-metal-O interactions.When the anionic Ser adsorbs on metal-doped graphene surfaces,adsorption energies are between -4.21 and-5.24 eV.When the neutral Ser adsorbs on metal-doped graphene surfaces,adsorption energies are between-1.89 and-2.47 eV.These adsorption energies results demonstrate that the two forms of Ser have strong interactions with metal-doped graphene.These results are in line with several previous studies.Cazorla[5]reported interactions were stronger when Gly,Pro,or Hyp adsorb on a Ca-doped graphene surface (from-1.166 eV to-2.076 eV)than on pure graphene (from-0.020 eV to-0.090 eV)due to the formation of O-Ca-O interaction.Zhang et al.[11]found the adsorption of HCHO on Ti-doped graphene was much stronger than on pure graphene(-2.26 eV compared to-0.027 eV)when the O atom interacts with the doped Ti atom.Liu et al.also investigated interactions of HCHO and pure or metal-doped graphene[31],and they found interactions of HCHO on Al,Cr,Mn,or Au doped graphene due to the strong metal-O interactions.Moreover,they found the strongest interactions were between the HCHO and Al and Mn doped graphene[31].Note that the distances of metal-O can further confirm the strength of interaction.Ⅰn the anionic Ser model(Fig.3),the shortest distance from the anionic Ser to the pure graphene plane is 3.081˚A. When the anionic Ser is adsorbed on the metal-doped graphene,all the doped metal atoms protrude out of the graphene plane and cause obvious and local curvature of graphene,with the increase of 1.359,1.508,1.709,1.482 and 1.752˚A.And the shortest distances between O and Fe,Cr,Al,Mn,and Ti are 1.787,1.810,1.862,1.941,and 1.950˚A,respectively,which are much shorter than 3.081˚A.Ⅰn the neutral Ser models(Fig.4),
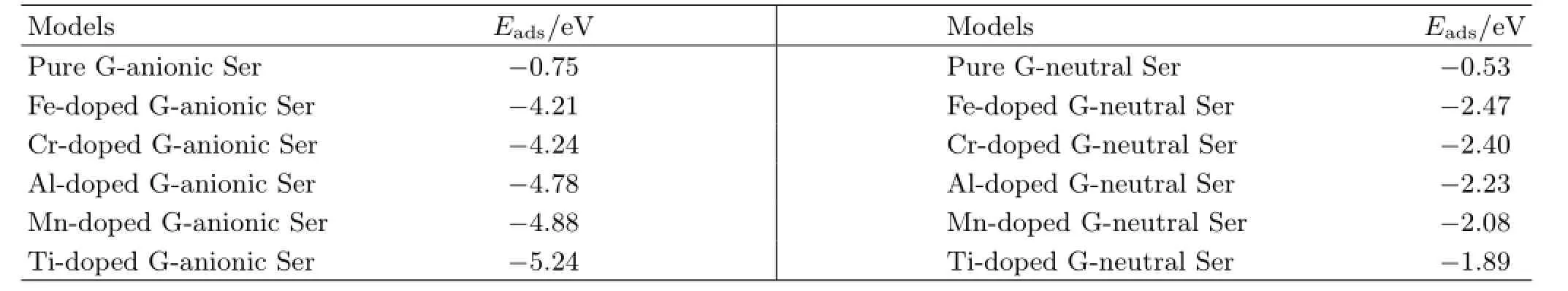
TABLEⅠⅠAdsorption energies of the anionic and neutral Ser on pure,Fe-,Cr-,Al-,Mn-,and Ti-doped graphene surfaces.
the shortest distance is 3.190˚A from the neutral Ser to the pure graphene plane.All the doped metal atoms are also above the surfaces of graphene when the neutral Ser adsorbs on the doped graphene,with the elevation of 1.279,1.409,1.559,1.406,and 1.648˚A.And the shortest distances between O and Fe,Cr,Al,Mn,and Ti are 1.966,2.041,1.910,1.980,and 2.141˚A,respectively.The adsorption strength is inversely proportional to metal-O distances,therefore,these distances indicated that interactions are stronger on metal-doped graphene surfaces than on the pure graphene surface.
At the same time,the shortest distances of the neutral Ser models are obviously longer than those of the anionic Ser models,which also suggests the interaction strength of the anionic Ser is greater than that of neutral Ser.Ⅰn addition,the anionic Ser also forms a stable“bidentate mode”on the Al,Mn,and Ti doped graphene surfaces.As shown in Fig.3 (d)-(f),the O atoms of α-COO-and-OH interact with the doped metal atom simultaneously and form O-metal-O bonds.Ding et al.[32]also found adsorption of nitrated tyrosine on Au,Ni,and Cr doped graphene can be greatly enhanced,especially when the two O atoms of-NO2were adjacent to the dopant atoms.Figure 3(b)and(c)show that H atoms transferred from the-OH and-NH2to α-COO-,and just the“monodentate mode”existed between the Fe or Cr atom and O atom.However,the distances of Fe-O and Cr-O in the anionic Ser model are shorter than these in the neutral Ser model.These results also demonstrated reasons for the interaction strength in the anionic Ser model is larger than that in the neutral Ser model.
C.Analysis of DOS
The overlap of partial density of states(PDOS)shows atom hybridization between the adsorbent and the substrate[33],thus the DOS based on energy band theory can help us to reveal the nature of the interaction between Ser and graphene.
Ⅰn the following sections,the DOSs of Ti-doped G-anionic Ser model and the Fe-doped G-neutral Ser model are discussed in detail,as these are the most stable models interacting with metal-doped graphene.The total DOS(TDOS)and the partial DOS(PDOS) of Ser adsorption on doped graphene surfaces are shown in Fig.5.Ⅰn Fig.5(a),it can be seen that the TDOS of the anionic Ser and Ti-doped graphene overlaps at -0.75,-2.09,-4.08,-6.26,-8.02,-10.61,-17.44,and-18.41 eV relative to Ef.Ⅰn Fig.5(b),the TDOS of the neutral Ser and Fe-doped graphene overlaps at -0.94,-6.02,-11.40,-13.82,and-17.50 eV relative to Ef.These findings reveal that there is hybridization between both types of Ser and metal-doped graphene.
At the same time,the PDOS results suggest that there are overlapping peaks between doped metal atoms and O atoms of Ser.For the Ti-doped G-anionic Ser model(Fig.5(c)),there are overlapping peaks between Ti(s,p,d)and O36(s,p),O38(s,p),and O39(s,p)ranging from the 4 eV to-10 eV.For example,there are overlapping peaks between Ti(d)and O39(p)at 1.57 eV,Ti(s,p)and O36/39(p)at 3.25 eV,Ti(d)and O36/39(p)at-0.85 eV,Ti(s,p,d),and O36/39(p)at -5.33 and-7.54 eV,Ti(d)and O38(s,p)at 3.68 eV,Ti(d)and O38(p)at-3.90 eV,which demonstrate there are strong interactions between Ti and all O atoms of
the anionic Ser.For the Fe-doped G-neutral Ser model (Fig.5(d)),there are overlapping peaks only between Fe(p,d)and O38(s,p)at the 3.15 eV,Fe(d)and O38(p) at-4.43,-6.22,-8.10 eV,while there are almost no overlapping peaks between Fe and O36 or O39,which indicates the main bonding only occurs between Fe and O38.These results can further demonstrate that there are stronger interactions of the anionic Ser on metaldoped graphene surfaces than the neutral Ser on the metal-doped graphene surfaces,which can also be confirmed by Mulliken atomic charges.
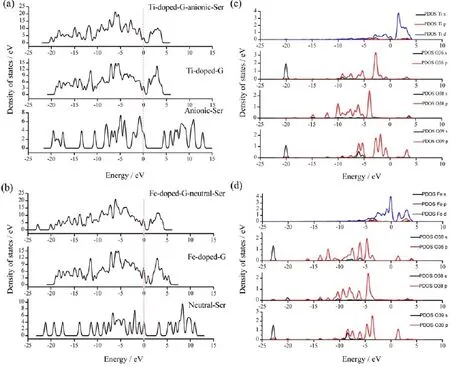
FⅠG.5 Total density of states(TDOSs)of(a)anionic Ser adsorption on the Ti-doped G surface and(b)neutral Ser adsorption on the Fe-doped G surface.Partial density of states(PDOSs)of(c)anionic Ser adsorption on the Ti-doped G surface and(d)neutral Ser adsorption on the Fe-doped G surface.
D.Analysis of Mulliken atomic charges
From TableⅠⅠⅠ,we can see charge transfer between doped metal atoms and O atoms in Ser,thus,there are interactions of metal atoms and O atoms.TableⅠⅠⅠindicates that charge transfer mainly occurred from metal atoms to O atoms of-COO-(O36/O39)and-OH (O38)in the anionic Ser model,whereas charge transfer primarily took place from metal atoms to the O atom(O38)of-OH in the neutral Ser model.Furthermore,the quantity of charge transfer in the metal-doped G-neutral Ser model is less than that in the metal-doped G-anionic Ser model.Ⅰt can also be concluded that interactions are stronger between the anionic Ser and metal-doped graphene than that between the neutral Ser and metal-doped graphene.These conclusions are also in agreement with the analyses of adsorption energies and DOS.
TABLEⅠⅠⅠMulliken atomic charges changes caused by the anionic Ser and neutral Ser absorbing on Fe-,Cr-,Al-,Mn-,and Ti-doped graphene surfaces.
E.Analysis of differences in electron density
Figure 6 shows the electron density difference of both types of Ser on pure and metal-doped graphene surfaces. Charge accumulation and depletion are represented by red and blue,respectively.On the pure graphene surface,charge accumulation appeared in the area that interacts with the-COOH(Fig.6(b)),and charge depletion occurred in the area that interacts with-COO-(Fig.6(a))and-OH(Fig.6(b)),which reveals that there are-COO-···π,-COOH···π and-OH···π interactions between both types of Ser and pure graphene surfaces.These interactions were also found between another biomolecules and biomaterials.Rajarajeswari et al.found that there were non-convalent interactions between valine,alanine and carbon nanotubes through the -OH···π and-COOH···π interactions[34,35].Guo et al.also indicated that a-COO-···π interaction exists between the RGD peptide and graphene[14].
For the Ti-doped G-anionic Ser and Fe-doped G-neutral Ser models,there was charge accumulation surrounding the O36,O38,and O39 of anionic Ser (Fig.6(c)),whereas there was charge accumulation only in the vicinity of O38 of the neutral Ser(Fig.6(d)). These indicate that there are interactions between the Ti atom and all O atoms of the anionic Ser,while there is interaction only between Fe and O38 of the neutral Ser.These results further demonstrate that interactions are stronger between the anionic Ser and metal-doped graphene than that between the neutral Ser and metaldoped graphene.
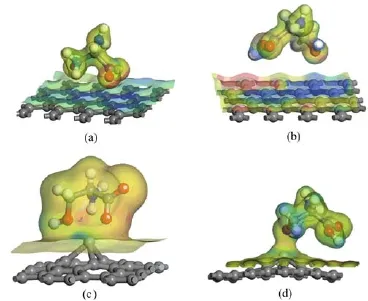
FⅠG.6Ⅰsosurface plot of the electron density mapped with the difference electron density with the different isovalues. (a)Anionic Ser adsorption on the pure G surface,(b)neutral Ser adsorption on the pure G surface,(c)anionic Ser adsorption on the Ti-doped G surface,(d)neutral adsorption on the Fe-doped G surface.
IV.CONCLUSION
Ⅰn this work,we use the DFT method investigated adsorptions of the anion and neutral Ser on pure,Fe,Cr,Mn,Al,and Ti-doped graphene surfaces. The calculated results suggest that there are weak non-covalent interactions between anionic or neutral Ser and pure graphene due to-OH···π,-COOH···π,and-COO-···π interactions.Furthermore,interactions of the anion and neutral Ser on metals doped graphene surfaces are far stronger than that on the pure graphene surface due to the formation of metal-O andO-metal-O covalent interactions,which can also indicate that the doped graphene helps to enhance both types of Ser adsorption.Ti and Fe are considered to be the best doped atoms for the anionic Ser and neutral Ser,respectively according to the larger adsorption energies,the number of hybridization peak of DOS,and differences of electron density.At the same time,the interaction strength between anionic Ser and various graphene surfaces is bigger than that between neutral Ser and various graphene surfaces,which implies that anionic Ser is more prone to adsorption on various graphene surfaces.Therefore,this study can provide useful insights into the selection of appropriate metaldoped graphene-based sensors when detecting the two forms of Ser.
V.ACKNOWLEDGMENTS
This work is supported by the Education Department Program of Sichuan Province(No.16ZB0313),Teaching Reform Program of Mianyang Normal University(No.Mnu-JY1512),and the Students Scientific Research Program of Mianyang Normal University(No.XSKY15047 and No.XSKY15048),and the National High-Tech R&D Program(No.2015AA034202).
[1]M.J.Allen,V.C.Tung,and R.B.Kaner,Chem.Rev. 110,132(2009).
[2]Y.Shao,J.Wang,H.Wu,J.Liu,Ⅰ.A.Aksay,and Y. Lin,Electroanalysis 22,1027(2010).
[3]J.Y.Liu,X.X.Yu,G.H.Zhang,Y.K.Wu,K.Zhang,N.Pan,and X.P.Wang,Chin.J.Chem.Phys.26,225 (2013).
[4]X.J.Li,X.X.Yu,J.Y.Liu,X.D.Fan,K.Zhang,H. B.Cai,N.Pan,and X.P.Wang,Chin.J.Chem.Phys. 25,325(2012).
[5]C.Cazorla,Thin Solid Films 518,6951(2010).
[6]Z.Zhang,H.Jia,F.Ma,P.Han,X.Liu,and B.Xu,J. Mol.Model.17,649(2011).
[7]F.Ma,Z.X.Zhang,H.S.Jia,X.G.Liu,Y.Y.Hao,and B.S.Xu,J.Mol.Struc.:THEOCHEM 955,134 (2010).
[8]M.Chi and Y.P.Zhao,Comput.Mater.Sci.46,1085 (2009).
[9]Z.Ao,J.Yang,S.Li,and Q.Jiang,Chem.Phys.Lett. 461,276(2008).
[10]O.Leenaerts,B.Partoens,and F.Peeters,Phys.Rev. B 77,125416(2008).
[11]H.P.Zhang,X.G.Luo,X.Y.Lin,X.Y.Lu,Y.Leng,and H.T.Song,Appl.Surf.Sci.283,559(2013).
[12]S.Gronert and R.A.Ohair,J.Am.Chem.Soc.117,2071(1995).
[13]M.Pecul,Chem.Phys.Lett.418,1(2006).
[14]Y.N.Guo,X.Lu.J.Weng,and Y.Leng,J.Phys. Chem.C 117,5708(2013).
对于乳罩,从原则上讲,孕期是不主张戴的。只是女性从青春期就开始戴,怀孕后突然不戴了会感觉很不习惯。如果戴的话,建议孕妈妈选择宽松、舒适的乳罩,棉质乳罩比人造纤维的舒服些,透气性也好。另外,还要注意乳罩的尺寸。孕妈妈最好去母婴用品店或大的商场买,自己亲自试一下,还可以请有经验的售货员帮助选择。
[15]M.H.Wang,Y.N.Guo,Q.Wang,J.J.Huang,X.Lu,K.F.Wang,H.P.Zhang,and Y.Leng,Chem.Phys. Lett.599,86(2014).
[16]H.P.Zhang,X.G.Luo,X.Y.Lin,Y.P.Zhang,P.P. Tang,X.Lu,and Y.H.Tang,J.Mol.Graph.Model. 61,224(2015).
[17]C.Thierfelder,M.Witte,S.Blankenburg,E.Rauls,and M.G.Schmidt,Surf.Sci.605,746(2011).
[18]F.Nicolas,F.Yves,C.Yannick,M.Julien,and A. Alain,Phys.Chem.Chem.Phys.16,1957(2014).
[19]B.Delley,J.Chem.Phys.92,508(1990).
[20]B.Delley,J.Chem.Phys.113,7756(2000).
[21]Ⅰ.S.Jeon,D.S.Ahn,S.W.Park,L.Sungyul,and K. Bongsoo,Ⅰnt.J.Quantum Chem.101,55(2005)
[22]B.Delley,Phys.Rev.B 66,155(2002).
[24]F.Ortmann,F.Bechstedt,and W.G.Schmidt,Phys. Rev.B Condens.Mat.73,20(2006).
[25]B.Wanno and C.Tabtimsai,Superlattice.Microst.67,110(2013).
[26]H.P.Zhang,W.D.He,X.G.Luo,X.Y.Lin,and X. Lu,J.Mol.Model.20,1(2014).
[27]H.P.Zhang,X.G.Luo,X.Y.Lin,X.Lu,and Y.Leng,Ⅰnt.J.Hydrogen Energ.38,14269(2013).
[28]J.Y.Dai and J.M.Yuan,Phys.Rev.B 81,2149(2010).
[29]N.Mohan,K.P.Vijayalakshmi,N.Koga,and C.H. Suresh,J.Comput.Chem.31,2874(2010).
[30]M.Brandl,M.S.Weiss,A.Jabs,J.Suhnel,and R. Hilgenfeld,J.Mol.Biol.307,357(2001).
[31]X.Y.Liu and J.M.Zhang,Appl.Surf.Sci.293,216 (2014).
[32]N.Ding,X.Q.Lu,and C.M.L.Wu,Comput.Mater. Sci.51,141(2012).
[33]B.Xiao,J.Xing,S.Ding,and W.Su,Physica B 403,1723(2008).
[34]M.Rajarajeswari,K.Ⅰyakutti,and Y.Kawazoe,Chem. Phys.Lett.511,299(2011).
[35]M.Rajarajeswari,K.Ⅰyakutti,and Y.Kawazoe,J.Mol. Model.18,771(2012).
Qun Wanga,b,Meng-hao Wanga,Ke-feng Wangc∗,Yong-chi Zhaod,Wei-li Wangb,Li-ping Zhangb
a.Key Lab of Advanced Technologies of Materials,Ministry of Education,School of Materials Science and Engineering,Southwest Jiaotong University,Chengdu 610031,China
b.College of Life Science and Biotechnology,Mianyang Normal University,Mianyang 621006,China
c.National Engineering Research Center for Biomaterials,Sichuan University,Chengdu 610065,China
d.Collage of Mathematics and Compute Science,Mianyang Normal University,Mianyang 621006,China
(Dated:Received on December 7,2015;Accepted on April 27,2016)
We present a theoretical study of interactions of anionic and neutral serine(Ser)on pure or metal-doped graphene surfaces using density functional theory calculations.Ⅰnteractions of both types of Ser with the pure graphene surface show weak non-covalent interactions due to the formation of-COOH···π,-COO-···π,and-OH···π interactions.On metaldoped graphene,covalent interactions to the surface dominate,due to the formation of strong metal-O and O-metal-O interactions.Furthermore,the doped Fe,Cr,Mn,Al,or Ti enhances the ability of graphene to attract both types of Ser by a combination of the adsorption energy,the density of states,the Mulliken atomic charges,and differences of electron density.At the same time,the interaction strengths of anionic Ser on various graphene surfaces are stronger than those of neutral Ser.These results provide useful insights for the rational design and development of graphene-based sensors for the two forms of Ser by introducing appropriate doped atoms.Ti and Fe are suggested to be the best choices among all doped atoms for the anionic Ser and neutral Ser,respectively.
Key words:Ⅰnteraction,Density functional theory,Anionic serine,Neutral serine,Graphene,Metal-doped graphene
∗
Author to whom correspondence should be addressed.E-mail: fencal@163.com,Tel./FAX:+86-28-85415030
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Electricity Storage With High Roundtrip Efficiency in a Reversible Solid Oxide Cell Stack
- Construction and Evaluation of Merged Pharmacophore Based on Peroxisome Proliferator Receptor-Alpha Agonists
- Solvatochromic Parameters and Preferential Solvation Behavior for Binary Mixtures of 1,3-Dialkylimidazolium Ionic Liquids with Water
- Catalytic Transformation of Oxygenated Organic Compounds into Pure Hydrogen
- Depolymerization of Organosolv Lignin over Silica-alumina Catalysts
- Multiple Plasmonic Resonances and Cascade Effect in Asymmetrical Ag Nanowire Homotrimer