蜂蜜中烟曲霉素残留的测定
2016-09-20曹葳蕤吴黎明中国农业科学院蜜蜂研究所农业部蜂产品质量检测中心北京100093
曹葳蕤 魏 月 吴黎明(中国农业科学院蜜蜂研究所 农业部蜂产品质量检测中心,北京100093)
蜂蜜中烟曲霉素残留的测定
曹葳蕤魏月吴黎明
(中国农业科学院蜜蜂研究所 农业部蜂产品质量检测中心,北京100093)
本文建立并优化了蜂蜜中烟曲霉素的HLPC-MS/MS分析方法。该方法的检出限为2 μg/kg,线性范围为1~1000 μg/L,R2为0.9992,在三个加标水平下,回收率在72.09~92.09%之间,批内和批间相对标准偏差分别在2.7~10.32%和5.2~11.1%。方法适用于蜂蜜中烟曲霉素残留的测定。
蜂蜜;烟曲霉素;分析方法
1 前言
近年蜜蜂微孢子虫在全世界流行,尤其是近十几年发现的东方蜜蜂微孢子虫,对蜂产业造成了严重损失[1]。烟曲霉素被认为是治疗蜜蜂微孢子虫病特效药[2],它是从烟曲霉菌(Aspergillus fumigatus)中分离的一种抗生素,具有突出的抗寄生虫效果[3]。然而由于知识产权、贸易保护等因素,蜂用烟曲霉素还没有在我国授权使用。为了争取烟曲霉素能被我国农业兽药管理部门允许在蜂群中使用,充分发挥其在治疗日益严重的蜜蜂微孢子虫病中的作用,避免甲硝唑等禁用药物的使用,有必要开展烟曲霉素在蜂产品中代谢降解规律等基础性研究。
关于烟曲霉素残留的分析方法已有一些报道[4-8],主要有酶联免疫法、高效液相色谱串联质谱法(HPLCMS/MS),液相色谱质谱法(HPLC/MS),高效液相色谱法(HPLC)。其中高效液相色谱串联质谱(HPLC-MS/MS)结合了高效液相色谱和串联质谱的优点,分离能力强、选择性好、灵敏度高,能达到更好检出限,其多反应监测扫描模式 (Multi-reactions monitoring,MRM)的选择性高。因此本文选择高效液相色谱串联质谱法对蜂蜜中的烟曲霉素进行研究。
本实验优化了蜂蜜中烟曲霉素的提取和HPLCMS/MS分析条件,建立了适用于高通量分析蜂蜜中的烟曲霉素残留的测定方法。为研究烟曲霉素在蜂巢体系和蜂产品中的变化规律研究提供了方法支持。
2 材料、试剂与仪器
2.1主要材料、试剂
蜂蜜,北京香山地区产
C18固相萃取小柱(6 ml/500 mg,美国Waters公司);
Oasis HLB固相萃取柱(6 ml/500 mg,美国Waters公司);
QuEChERS试剂盒(美国Agilent公司);
Zorbax SB-C18色谱柱(50 mm×2.1 mm,1.8 μm);
Zorbax XDB-C18色谱柱(50 mm×2.1 mm,1.8 μm);
Poroshell 120SB-C18色谱柱 (100 mm×2.1 mm,2.7 μm);
甲醇、乙腈、甲酸、乙酸乙酯均为色谱纯(美国MREDA technology公司);
水为超纯水,用milli-Q超纯水系统制备(美国Millipore公司)。
2.2标准品
烟曲霉素标准品CSA:23110-15-8,1 mg,纯度98%(北京百灵威科技有限公司),用1 ml甲醇溶解,配制成浓度为1.0 mg/ml的烟曲霉素贮备液,保存于-20℃冰箱中备用。
2.3仪器与设备
Agilent 6460高效液相色谱串联质谱仪(美国安捷伦公司);
固相萃取装置(美国Waters公司);
CR22GⅡ高速冷冻离心机(日本日立公司);
N-EVAP112氮气吹干仪(美国OA-SYS公司);
QB-210试管翻转混合器(海门市其林贝尔仪器公司);
Milli-Q纯水机(美国Millinpore公司)。
2.4标准溶液的制备
用甲醇稀释将浓度为1.0 mg/ml烟曲霉素标准储备液稀释成1000 μg/L、500 μg/L、200 μg/L、100 μg/L、10 μg/L 和1 μg/L的工作液。
2.5色谱条件和质谱条件
2.5.1高效液相色谱条件
色谱柱:PoroshellSB-C18柱(2.1×100 mm,2.7μm);
流动相:0.1%甲酸水溶液(A)+乙腈(B);
流速:0.2 ml/min;
梯度:0min,45%B;3~6.5min,90%B;6.6~15 min,45%B;
柱温:30℃;
进样量:5 μl。
2.5.2质谱条件
电喷雾离子源(ESI),正离子扫描模式,检测方式采用多反应监测(MRM)模式,气体温度(Gas Temp)300℃,气体流速(Gas Flow)6 L/min,喷雾器压力(Nebulizer)45 psi,鞘气温度(Sheath Gas Temp)350℃,鞘气流速(Sheath Gas Flow)9 L/min,毛细管正电压3500 v。
2.6样品处理方法
取5 g蜂蜜样品用10 ml纯水溶解,8000 r/min离心5 min。吸取上清液于Oasis HLB固相萃取柱(使用前用5 ml甲醇和5 ml纯水活化)中进行净化。样品溶液全部流出后,用10 ml纯水淋洗HLB小柱,负压下抽干,用6 ml乙酸乙酯洗脱,收集洗脱液,40℃下氮气吹干,用1ml甲醇复溶,将溶液过0.22 μm尼龙滤膜后待测。对于超过线性浓度样品,稀释后重新测定。
3 结果和讨论
3.1仪器条件的优化
3.1.1色谱柱的选择
由图1烟曲霉素的结构可知,烟曲霉素是一种弱极性物质,一般采用反相色谱柱对其进行分析。文献报道中一般采用苯基柱和C18柱等反相色谱柱来分离[6]。本研究对三种不同的色谱柱进行比较:Zorbax SB-C18(50 mm×2.1 mm,1.8 μm),Zorbax XDB-C18(50 mm×2.1 mm,1.8 μm),Poroshell 120,SB-C18(2.7 μm,100 mm× 2.1 mm)。如图Poroshell 120,SB-C18的可以得到满意分离效果、较高灵敏度以及良好的峰型。最终,选择Poroshell 120,SB-C18作为本次试验的色谱柱。
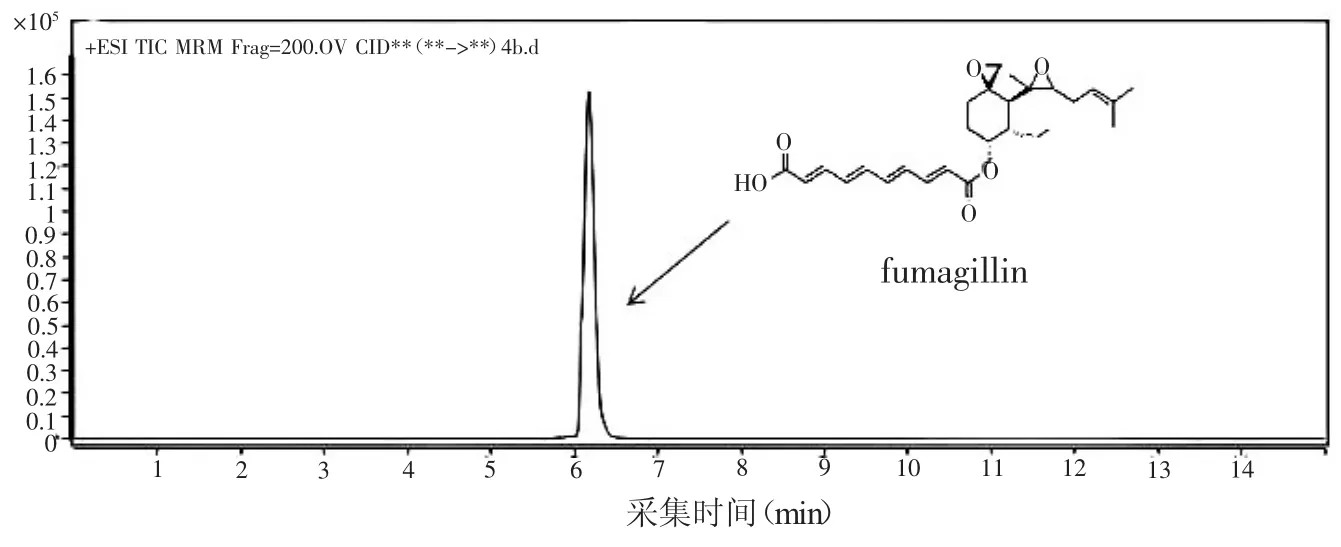
图1 烟曲霉素标样的TIC图
3.1.2流动相及洗脱梯度的优化
为了得到最佳的色谱分离效果,提高烟曲霉素测定的灵敏度,本研究对流动相和添加物进行了优化。首先对流动相的组成进行了优化,比较了使用纯水-乙腈,纯水-甲醇,0.1%甲酸水-乙腈,0.1%甲酸水-甲醇作为流动相后,色谱峰的保留时间与峰形。结果表明,乙腈作为有机相,其响应值比甲醇高,出峰时间早,这可能是由于乙腈的极性比甲醇强的缘故。流动相中分别加入0.05%、0.1%、0.5%的甲酸,发现0.1%的甲酸可以明显提高正模式下母离子加氢峰的强度。
然后对洗脱梯度进行优化,最终确定流动A为0.1%甲酸水溶液,B相为乙腈。梯度洗脱程序:0~3 min,45%B;3~6.5 min,90%B;6.6~15 min,45%B。在该梯度洗脱程序下,峰形尖锐,响应较高。
3.1.3质谱条件的优化
为了得到最好的定量响应,首先将烟曲霉素的标准溶液(1.0 μg/ml),通过电喷雾离子化直接进入质谱进行一级全扫描分析,分别尝试了三种模式:正模式下加酸(ESI+),负模式下加酸(ESI-)以及负模式下不加酸,发现在正模式加酸的条件下得到的响应值最高,因此选择正离子监测模式,得到其稳定的[M+H]+和碎片。在此过程中对一些质谱参数进行优化,确定最优的碎裂电压(Fragmentor Voltage)、碰撞能量(Collision Energy)等,以获最大灵敏度。然后在多反应监测模式(MRM)下对其碎片离子进行全扫描,如图2,得到了4个峰值较大的离子碎片∶m/z427,m/z265,m/z232,m/ z176,满足欧盟指令(2002/657/EC)中利用质谱方法对药物残留进行分析确证必须满足4个识别点的要求[9]。由于m/z 427是烟曲霉素脱水形成的 [M-H2O]+,不稳定,m/z232峰度比较小,因此选择质荷比(m/z)分别为265.1和 176.9,其中 m/z 179.6作为定量离子,m/z 265.1作为定性离子。
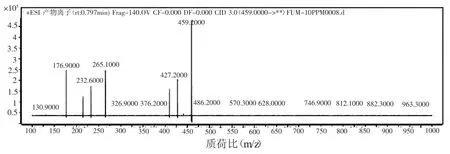
图2 烟曲霉素全扫描质谱图
3.2前处理方法优化
3.2.1QuEChERS方法与固相萃取方法的选择
选择合适的萃取方式及萃取柱是决定固相萃取效率的关键因素,可以实现对样品的净化浓缩,降低基质的干扰,从而改善待检测物质的离子化程度,提高检测方法的选择性和灵敏度。
基质效应是指出目标物质外的其他成分对于定量分析的综合影响,一般认为是待测组分与样品基质成分在雾滴表面离子化过程中存在竞争所导致,会对目标物的离子化效率,使离子强度增加或降低,从而影响检出限、定量限以及测定结果的准确性[10]。本研究中向空白基质中添加一定浓度的烟曲霉素标准品,通过比较实际测定浓度与所添加浓度的比值来评价基质影响。若比值大于1,则基质效应表现为基质增强(用“+”表示),若比值小于1,则表现为基质抑制(用“-”表示)。一般情况下,基质影响可接受范围为-20%~10%。
本文章比较了QuEChERS方法与固相萃取方法(Oasis HLB、C18)的提取效果。称取5.0 g蜂蜜溶于10 ml去离子水中,配制成0.5 g/ml的蜂蜜溶液作为样品溶液。样品溶液平行准备3组,每组3份。分别采用上述三种方法对3组蜂蜜样品进行前处理后,向每组的3份平行的蜂蜜样品中分别添加10、50、100 μg/L的标准溶液,进行后续实验操作,对比四种方法的回收率及基质效应。结果见表1。
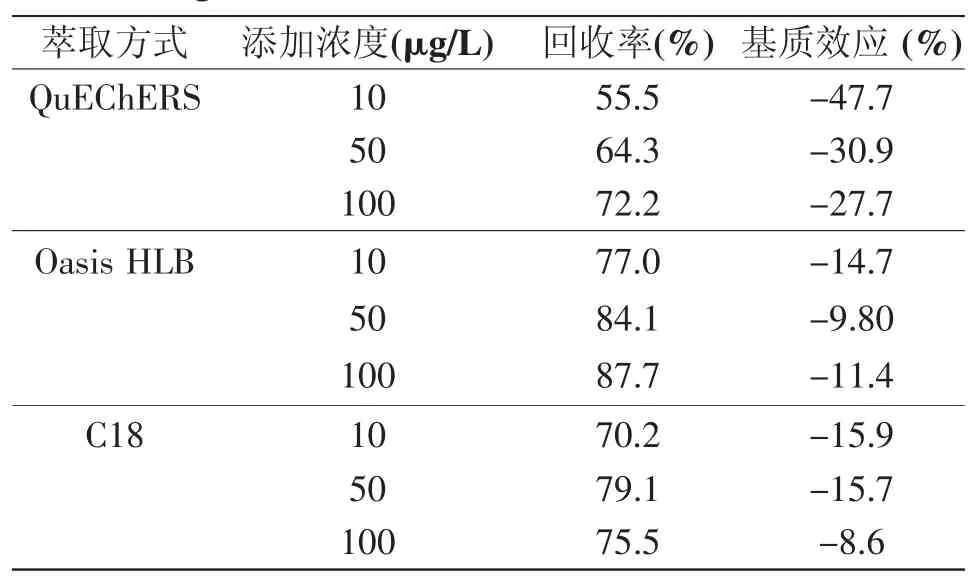
表1 QuEChERS、Oasis HLB、C18提取结果对比
由表1可知,三个方法都表现为基质抑制。采用QuEChERS方法对目标物进行提取,回收率较低,且基质效应严重,低浓度添加水平时基质影响尤为突出,严重干扰了测定结果的准确性。而且,如果研究样品批次多、样品量大,若对每批次样品分别进行基质校正,会影响检测结果的重复性。QuEChERS方法虽然具有快速、简单的优势,更适用于目标物质的快速筛查。烟曲霉素降解代谢等规律的研究需要对目标物质准确定量,因此QuEChERS方法并不适用。
由表1可知,采用固相萃取方法的Oasis HLB和C18柱子提取烟曲霉素时,净化效果好,回收率均高于QuEChERS方法,没有明显的基质效应,因此本研究选择固相萃取作为提取方法。由结果可知,Oasis HLB柱子在三个添加浓度下的回收率均高于C18柱,对目标物的保留能力较C18柱高。因此,本研究选取HLB柱子对蜂蜜中的烟曲霉素进行提取。
3.2.2淋洗溶液和洗脱溶液的选择
参考相关文献[10]淋洗溶液对纯水、2%的甲醇、10%的氨水进行比较,由图3可知,纯水的淋洗对回收率的损失较小,效果最好。
本研究对洗脱溶液(甲醇、乙腈、乙酸乙酯)进行了筛选,并对洗脱液体积进行了优化。对比甲醇、乙腈、乙酸乙酯的洗脱效果可知,甲醇、乙腈洗脱物较多,测定时存在杂质干扰,且目标物回收率低。乙酸乙酯洗脱物种类少、洗脱效果好,目标物质回收率高,因此实验中选择乙酸乙酯作为洗脱溶液。分别采用2、3、4、5、6、7、8、9、10 ml乙酸乙酯对固相萃取柱进行洗脱,对比回收率,结果见图4,洗脱液体积由2 ml增加至6 ml时,回收率逐渐升高,此后体积增加,回收率无明显变化,说明6 ml乙酸乙酯已洗脱完全,因此,本实验选取6 ml乙酸乙酯作为洗脱溶液。
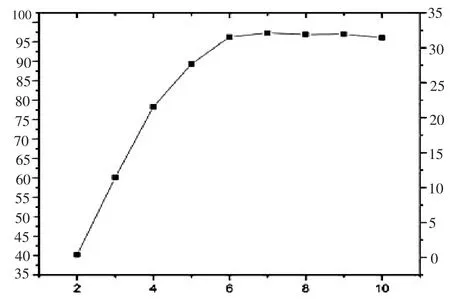
图3 淋洗溶液的优化
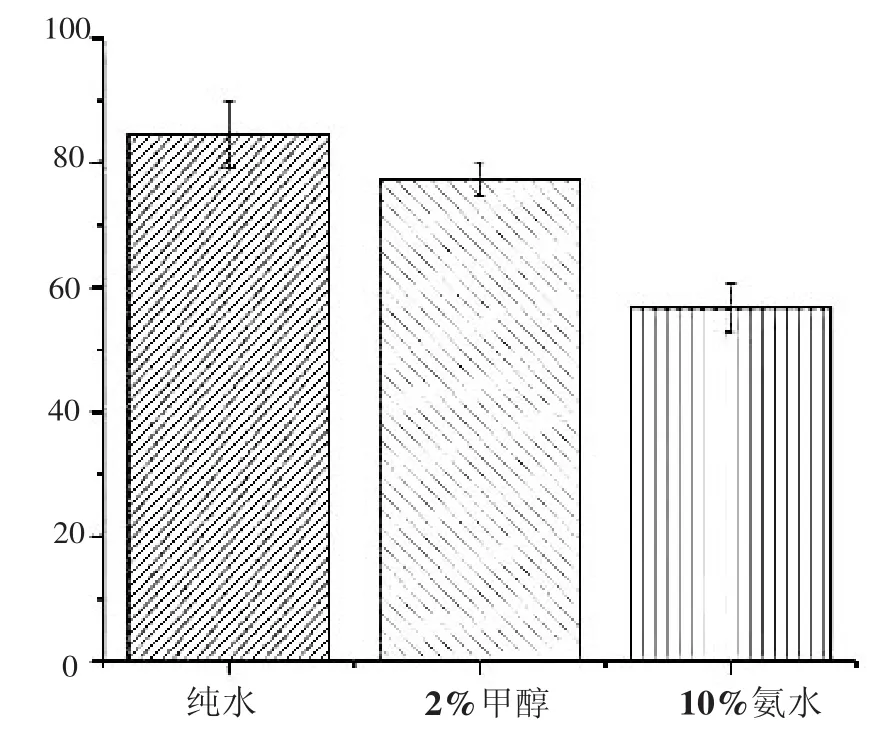
图4 洗脱溶液体积的优化
3.3方法评价
3.3.1线性范围及检出限
以水为溶剂稀释烟曲霉素储备液,配制质量浓度为1 μg/L、10 μg/L、100 μg/L、500 μg/L、1000 μg/L的标准系列标准溶液,按照2.4中优化的实验条件进行测定。以定量离子的峰面积(y)为纵坐标、质量浓度(x)为横坐标,绘制标准曲线。结果表明,线性范围在1~1000 μg/ml之间(相关系数R2=0.9992,线性方程y=13.087x-607.94)。配制接近方法检出限的标准样品,重复测定7次,以信噪比为3时对应的质量浓度作为的方法的检出限,为2.0 μg/kg,与以往研究相比,灵敏度更高。
3.3.2方法的添加回收率、精密度
空白蜂蜜样品中添加一定浓度烟曲霉素储备液,分别制成5 μg/kg、10 μg/kg和50 μg/kg,每个加标水平做5个重复,连续做3 d,蜂蜜中烟曲霉素加标回收率、批内精密度和批间精密度结果见表2。三个水平添加回收率分别为 80.37~92.09%,78.51~90.32%,72.09~81.86%,日内精密度分别为5.6%、2.7%、10.3%,日间精密度分别为9.5%、5.2%、11.1%,均小于20%。
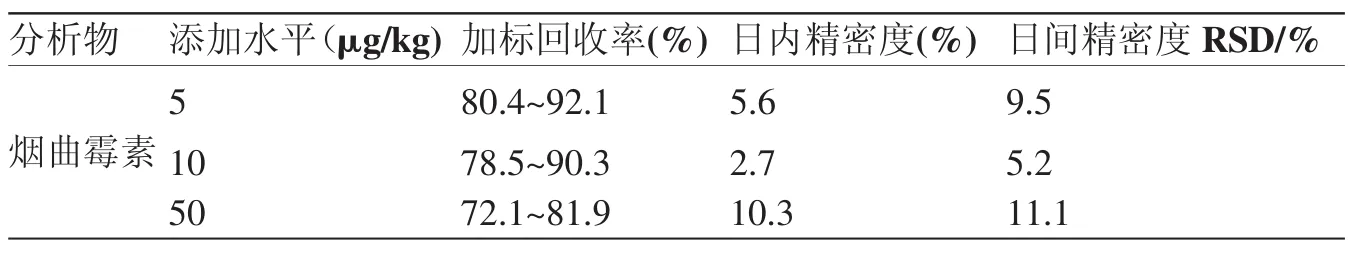
表2 烟曲霉素的添加回收率、精密度
4 结论
本文采用高效液相色谱-串联质谱(HPLC-MS/ MS)技术及固相萃取技术,建立并优化了蜂蜜烟曲霉素残留的检测分析方法。经方法学验证:烟曲霉素在1~1000 μg/L范围内,均具有良好的线性关系,线性相关系数(r)为0.9992,本文建立的检测方法检出限为2μg/ kg。在5 μg/kg、10μg/kg和50μg/kg三个加标水平下,回收率在72.09~92.09%之间,批内相对标准偏差在2.7~10.32%之间,批间相对标准偏差在5.2~11.1%之间。本方法操作简单、灵敏度高适用于大批量蜂蜜中烟曲霉素残留检测。
[1]Meghan OM,Toan T,WeiFong H,et al.Comparative virulence and competition between Nosemaapis and Nosemaceranae in honey bees (Apis mellifera).Journal of Invertebrate Pathology.2015,125(2): 9-15.
[2]Huang WF,Solter L,Yau P,et al.Effects of Fumagillin on Nosemaceranae Infections in Honey Bees(Apismellifera)[J]. American Bee Journal,2013,153(8):885-886.
[3]McCowen M C,Callender ME,Lawlis J F.Fumagillin(H-3),a newantibioticwithamebicidalproperties.Science,1951,113,202-203.
[4]Lopez MI,Pettis JS,Smith IB,et al.Multiclass determination and confirmation of antibiotic residues in honey using LC-MS/MS[J]. Journal ofAgricultural and Food Chemistry,2008,56(5):1553-1559.
[5]Kanda M,Sasamoto T,Takeba K,et al.Rapid determination of fumagillin residues in honey by liquid chromatography-tandem mass spectrometry using the QuEChERS method[J].Journal of AOAC International,2011,94(3):878-899.
[6]Dmitrovic J,Durden DA.Analysis of fumagillin in honey by LC-MS/MS[J].Journal ofAOACInternational,2013,96(3):687-695.
[7]Assil HI,Sporns P.ELISA and HPLC methods for analysis of fumagillin and its decomposition products in honey[J].Journal of Agricultural and Food Chemistry,1991,39(12):2206-2213.
[8]Nozal MA,Bernal JL,Martin MA,et al.Trace analysis of fumagillin inhoneybyliquidchromatography-diodearray-electrospray ionization mass spectrometry[J].J Chromatogr A,2008,1190(1-2): 224-231
[9]Commission Decision 2002/657/EC of the 12th August,2002 implementing Council Directive 96/23/EC concerningtheperformanceofanalytical methods and the interpretation of result,Official Journal,2002,L221:8
[10]许森.复杂基质样品中抗生素的实时直接分析质谱筛查技术研究[D].中国地质大学(北京),2015.
Determination of fumagillin residues in honey
Cao WeiruiWei YueWu Liming
(Institute of Apicultural Research,Chinese Academy of Agricultural Sciences Bee Product Quality Supervision and Testing Center,Ministry of Agriculture,Beijing 100093,China)
Abstract∶A method was developed to determinate fumagillin in honey using HPLC-MS/MS.The method was validated∶limit of detection(LOD)was 2 μg/kg;R2was 0.9992 when the linear ranged from 1 to 1000 μg/L;and the recoveries were between 72.1%and 92.1%with the intra-RSD of 2.7~10.3%and inter-RSD of 5.2~11.1%at three spiked levels.The developed method has been used to detect fumagillin residue in real samples.
honey,fumagillin,analytical method
吴黎明,E-mail:apiswu@126.com,Tel:010-62594643。
