植物关联分析方法的研究进展
2016-09-18冯建英温阳俊章元明
冯建英 温阳俊 张 瑾 章元明
1南京农业大学作物遗传与种质创新国家重点实验室, 江苏南京210095;2华中农业大学植物科技学院, 湖北武汉430070
植物关联分析方法的研究进展
冯建英1温阳俊1张瑾1章元明2,*
1南京农业大学作物遗传与种质创新国家重点实验室, 江苏南京210095;2华中农业大学植物科技学院, 湖北武汉430070
关联分析在人类和动植物遗传研究中的应用日益广泛, 新方法及其软件包不断涌现。为对其更好选择和应用, 本文综述了关联分析的主要方法及其软件包。首先, 介绍了群体结构对关联分析的影响; 其次, 重点介绍了单位点关联分析、多位点关联分析、上位性和多性状关联分析方法及其软件包; 最后, 展望了关联分析的发展动向。应当指出, 基于群体结构和多基因整体背景控制的全基因组单标记快速扫描算法在目前的实际资料分析中应用较广泛,与其结果互补的是假阳性率较高的非参数方法。但是, 今后的方法应当是以多位点模型、环境互作、上位性检验和多个相关性状联合分析为主。这为今后的理论与应用研究提供了有益信息。
全基因组关联分析; 上位性; 混合线性模型; 多位点模型
以分子标记与复杂性状基因间的连锁不平衡(linkage disequilibrium, LD)为基础的关联分析是人类复杂疾病遗传剖析的基本方法。近年来, 在动植物数量性状遗传分析中也有大量报道。
在人类复杂疾病的关联分析中, 最早分析的是家系资料和Case-Control数据。随着Risch提出了全基因组关联分析[1](genome-wide association study,GWAS), 特别是学者们研制了PLINK[2](http://pngu. mgh.harvard.edu/~purcell/plink/)和BOOST[3](http:// bioinformatics.ust.hk/BOOST.html)等应用软件包以后, 涌现出大量GWAS研究论文[4-6], 掀开了人类复杂疾病遗传研究和人类基因组学研究的新篇章。在植物遗传方面, Thornsberry等[7]在考虑群体结构的情况下用统计方法研究了玉米开花期的Dwarf8多态性变异, Hansen等[8]将GWAS用于海甜菜抽薹基因B的遗传研究, 特别是Zhang等[9]和Yu等[10]建立了混合线性模型关联分析方法以后, 学者们应用不同的统计模型和参数与非参数估计方法在全基因组开展了单标记扫描、快速计算算法、多位点模型、上位性检测和多个相关性状联合分析等大量研究。目前, 应用最多的是基于混合线性模型的快速计算方法, 它们大多是基于群体结构和多基因背景控制的单标记扫描方法, 例如, EMMA[11]、CMLM[12]及其拓展的算法。与此类方法互补的是非参数检验方法, 但是其假阳性率往往较高[13]。今后的方向是多位点模型、环境互作、上位性检验和多个相关性状联合分析的方法, 例如mrMLM[14]、FarmCPU[15]和QTXNetwork[16]。
植物复杂性状的关联分析方法已有很多, 各具特点, 各有所长。为增加这些方法的推广应用, 本文从统计模型、方法和应用条件上综述植物关联分析方法与软件研究的进展, 并展望今后的发展趋势, 为应用工作者更好地选择和应用这些方法提供方便。
1 群体结构对关联分析的影响
关联分析的一个主要问题是因群体结构造成的目标性状与无关基因间的假关联, 导致关联分析的假阳性率较高。为此, 提出了基因组控制[17-18]、结构关联[19-21]、主成分分析[22]和多维尺度[23]来解决群体结构对关联分析的影响。实际上, 关联分析也受多基因背景效应的影响。Bulik-Sullivan等[24]认为,目前的方法不能有效区分群体结构和多基因背景效应对关联分析的影响。为此, 提出了LD (linkage disequilibrium)得分回归方法。
2 单位点关联分析
随着基因组测序技术的进步及其成本的下降,关联分析在作物遗传育种研究中的应用越来越广泛。目前, 已经涌现出大量的关联分析方法与软件包。
2.1混合线性模型(mixed linear model, MLM)方法
Zhang等利用品种系谱、分子标记信息和数量性状观察值首先提出了植物品种资源群体关联分析的混合线性模型方法[9], 其数学模型为


若品种系谱不全, 不能计算IBD值; 若系谱不准确, 计算结果不可靠。特别是, 群体结构也会导致假关联。为克服这些缺点, Yu等[10]利用分子标记信息计算品种间多基因亲缘关系矩阵K, 以代替多基因IBD矩阵, 并引入群体结构矩阵Q, 以检测与数量性状关联的分子标记, 通过模拟人类和玉米数据集, 证实了新方法在提高QTL检测功效和控制I型错误率的有效性。此后, 混合线性模型关联分析方法得到较大发展和广泛应用。
2.2快速检测方法
随着测序技术的进步和测序成本的下降, SNP(single nucleotide polymorphism)标记在植物关联分析中应用已成为常态。但是, 大量的SNP标记会使得分析的运算时间增加。所以, 新的快速检测方法越来越受到应用者的青睐。
上述混合线性模型方法需要估计3个方差组分,若标记数目较多, 则运算时间较长。为此, Zhang等[12]提出了压缩混合线性模型(compressed mixed linear model, CMLM)方法。与Yu等[10]方法比较, CMLM方法将QTL效应视为固定效应; 利用聚类分析方法将品种分组, 获得最优分组数, 用组间亲缘系数代替品种间亲缘系数; 提出了固定多基因方差与误差方差比值的P3D (population parameters previously determined)算法。由此, 提高了检测功效和节约了计算时间。若寻找8种聚类分析方法和3种组间亲缘系数算法的最优组合, 还可进一步改进CMLM方法的检测功效, 这就是优化压缩混合线性模型(enriched CMLM, ECMLM)方法[26]。
Kang等[11]将模型(1)中的QTL效应u视为固定效应, 并记, 则

这些方法的提出缓解了海量SNP关联分析计算复杂度高和计算速度慢的问题。Kang等[10]分析玉米和拟南芥数据后认为, EMMA方法可有效降低群体结构导致的高假阳性, 且检测结果具有更高的稳定性和精度。Zhao等[34]将EMMA方法用于全球28个国家413个水稻品种的关联分析, 建立了水稻全基因组关联分析的开放平台。此外, Wen等[35]利用EMMA和P3D方法进行了大豆猝死综合症(sudden death syndrome)的全基因组关联分析。
此外, 非参数方法在植物关联分析中也得以应用。针对关联作图群体数量性状表型分布不对称、QTN (quantitative trait nucleotide)效应中等和感兴趣的等位基因频率很低的具体情况, Yang等[36]将非参数Anderson-Darling检验应用于关联分析, 并通过IBD和K值邻近法补全缺失SNP标记信息, 分析17个玉米数量性状后认为, 所获结果与常用关联分析结果可相互补充, 有利于发现常规方法不易发现的显著QTN。
3 多位点关联分析
上述关联分析混合模型方法及其快速算法是基于群体结构和多基因背景控制的单标记分析。连锁分析表明, 多QTL定位是提高QTL检测功效与精确度的有效途径。因而, 多位点关联分析方法学研究一直备受关注。
3.1广义线性模型(generalized linear model,GLM)方法
广义线性模型的一般形式可表述为

其中, yi和ηi分别是第i个品种性状表型观察值和潜在变量值; 函数h(·)是连结函数(link function),h-1(·)是其逆函数; E(·)为数学期望; β0是包含群体均值和群体结构的固定效应向量; βj是第j个标记的效应, xij是相应的哑变量; εi是随机误差。
由于h(·)可将性状表型观测值yi与潜在变量ηi联系起来, 因此广义线性模型可以为数量性状和离散型抗性性状遗传分析提供新方法, 而且也可以处理误差εi非正态性的情形。McCullagh和Nelder[37]系统阐述了广义线性模型相关理论。这些理论与方法在生物和医学领域被广泛应用, 推动了数量遗传学的发展。
为了改善遗传分析效果, 可将效应βj视为概率密度函数f1(βj|a)的连续型随机变量, 而参数a为概率密度函数f2(a|b,c)的连续型随机变量, 其中b和c可以是人为给定, 也可以是未知随机变量。这些分层超参数(hierarchical hyperparameters)a、b和c由其后验分布参数确定。例如, Yi等[38]提出的复杂疾病稀有等位基因(rare allele)和位点间上位性互作检测的分层广义线性模型, Feng等[39]提出的品种群体抗性分级性状关联分析的分层广义线性模型方法,Wang等[40]为解决关联分析的通路(pathway)问题建立的基于BLUP估计的广义线性混合模型方法。
3.2Bayesian方法
Iwata等[41]提出了品种资源群体多QTL检测的Bayesian关联分析方法, 其统计模型为

其中,yi为数量性状观察值; qij为群体结构Q矩阵第i行第j列元素, αj是第j亚群效应; xik表示第i品种标记k的基因型值; 假定每个QTN都在标记上,γk是指示变量, 若第k个标记存在效应为βk的QTN,则γk=1, 否则γk=0; εi是服从正态分布)的误差。假定参数先验分布为、 γk~Beta(1,pk)和, 其中、pk、vε和都是超参数, α的先验分布为常数。由此, 推导出各参数的条件后验分布, 通过Markov链Monte Carlo方法, 得到各参数的估计值。通过模拟和水稻数据分析表明, 该方法假阳性率低, QTN效应估计值偏差较小, 但是收敛较慢, 计算时间较长。若只有数百个分子标记, 还是有实用价值的。Iwata等[42]将这种方法拓展至离散型抗性性状多QTL检测的Bayesian关联分析。其主要思想是利用阈模型将抗性性状观察值转换为潜在连续性变量。
为了缩短Bayesian方法的计算时间, Zhang和Xu[43]将Bayesian方法的先验分布密度函数与似然函数相结合构建惩罚似然函数, 对提出的惩罚最大似然方法可进行连锁分析。相似地, Hoggart等[44]提出了分析case-control数据的惩罚logistic回归方法。虽然两者都是利用惩罚似然函数来估计模型参数,但是前者针对连续性变量的连锁分析, 而后者是针对case-control数据的关联分析。若模型中变量个数不超过样本容量的10倍, 这两种方法是可行的。不过, 对检测小效应QTL的功效有待提高。
3.3混合线性模型方法
针对结构群体(structured population)复杂性状,Segura等[45]提出了一种多位点混合模型关联分析方法。它利用了向前和向后逐步回归, 在变量筛选的每一步都需要先估计多基因方差2 gσ和残差方差,由此获得每个SNP广义最小二乘效应的估计值及其概率; 将最显著的SNP作为协变量放入混合模型中,进行全基因组条件分析, 获得F测验的概率P值。重复这一过程先完成向前回归, 再进行向后回归变量筛选。在筛选变量过程中, 通过Gram-Schmidt算法提高运算速度。模拟研究证实, 它比单标记分析具有更高的检测功效和较低假阳率; 在人类和拟南芥实际数据分析中, 识别到了新的关联位点。Liu等[15]将固定模型与随机模型迭代使用提出的FarmCPU方法与Segura等的方法在思想上有些相似, 也能检测到更多的已知基因。应当指出, 它主要是利用bin的思想显著减少模型中变量个数, 并节省存贮空间。Yang等[46]提出的GCTA方法, 是通过一条染色体或整个基因组上的所有SNPs估计方差组分, 研究所有QTN对性状的影响。
目前, 在广泛应用的关联分析方法中, 多数是将SNP效应视为固定效应。然而, Goddard等[47]认为,将SNP效应视为随机更好, 可将与目标性状无关的SNP效应压缩至0, 让表型观察值与预测值达到最大相关。但是, 并未提供SNP效应估计方法。为此,Wang等[14]结合多位点模型、新的矩阵变换和快速计算算法提出了多位点随机SNP效应混合线性模型方法。由于多位点特性, 并不需要多重检验矫正。模拟研究表明, 它比EMMA方法的QTN检测功效更高, 效应估计值更准; 以拟南芥6个开花期数据分析表明, 它能检测出更多的已知基因。
3.4Bayesian方法与混合模型方法的有机融合
混合模型假设有大量的小效应QTN, Bayesian方法则假设有少量的大效应的QTN。Zhou等[48]认为, 在实际资料分析时, 无法判定哪一种更符合资料本身。由此, 建议将两种方法结合, 提出Bayesian稀疏混合模型方法。其方法是假设QTN效应kβ服从混合正态分布。若0=q, 就是混合模型方法; 若0=, 就是Bayesian方法。模拟研究发现, 新方法在单个QTN解释的表型变异估计方面兼备混合模型和Bayesian两种方法的优点, 在育种值预测方面优于两种方法。Moser等[49]提出了一种类似方法, 称为Bayesian混合分布模型方法。它假定SNP效应服从4个正态分布的混合分布, 且固定每个成分分布的相对方差, 即,其中混合比例ip有,是所有SNP解释的加性遗传方差。其目的是将基因检测、SNP贡献率估计、复杂性状遗传基础和表型值预测相结合。通过人类遗传疾病数据分析认为, 大于96%的SNP效应是微小的; 大效应位点解释表型方差的比例因性状而异; 预测分析证实, 分析大效应控制的性状时, Bayesian方法更优。
4 上位性与多性状关联分析
4.1上位性关联分析
上位性关联分析的研究更充实了数量遗传学内容。但是, 超饱和线性模型问题和大数据问题更为突出, 计算复杂度显著增加。目前的研究主要集中在人类遗传, 应用参数和非参数检测方法。
在参数方法方面, Zhang和Liu[50]利用Bayesian原理和Markov链Monte Carlo方法, 提出了casecontrol数据同时检测主效和上位性QTN的Bayesian上位性关联作图BEMA法, 以推断与疾病显著相关的SNP。Zhang和Liu[50]的模拟研究表明, 能处理10万个SNP, 提高QTN检测功效。Tang等[51]结合Bayesian标记剖分模型和Gibbs抽样提出了检测上位性QTN的方法。Cho等[52]提出了一种基于惩罚logistic模型的弹性网正则化方法, 通过变量筛选和弹性网两步实现了上位性关联分析。在非参数方法方面, Han等[53]提出了DASSO-MB算法; Han等[54]提出一种基于Markov链的上位性互作检测FEPI-MB算法, 减小了搜索空间, 运算速度更快, 检测功效高于BEMA方法。Li等[55]提出一种两步非参数独立筛选方法, 以鉴定与性状潜在关联的主效和上位性位点, 最后再用LASSO等惩罚回归分析获得与性状显著关联的主效与上位性位点。他们认为, 其模型更具一般性, 还可获得无主效应位点间的互作, 更好地揭示控制性状的基因网络。与Yang等[36]的方法相比, 其假阳性率低是由于在非参数方法基础上增加了压缩估计, 并能估计主效QTN和上位性互作的效应值。
在植物遗传方面, Wang等[56]提出以自适应混合LASSO方法检测上位性; Lü等[57]提出了上位性检测的经验贝叶斯方法; Zhang等[16]提出基于图形卡GPU计算的混合模型方法, 以检测主效、基因与环境和基因间互作的QTN, 大大提高了计算速度; Wen等[58]提出基于EBLASSO算法的上位性检测方法, 分析了部分NCII遗传交配群体不同遗传组分对杂种优势的贡献。前两方法的模型中包含的变量个数不宜大于样本容量的10倍; 后两种方种是动态地向模型中引入变量, 可以容纳更多的变量, 处理海量变量的问题。应当指出, 上位性关联分析方法还有待进一步探索, 以提高运算速度和小效应基因互作检测功效。
4.2多个相关性状的关联分析
单一育种目标已成过去, 高产、优质和多抗是当前的育种目标。为了将遗传分析与作物育种更紧密结合, 有必要进行多个相关性状联合的关联分析。最容易想到的是主成分分析[59]、典范相关分析[60]、多个依变数的线性回归分析[61]、Meta分析[62]和偏最小二乘法[63]。当然, 关联分析最常用的还是混合模型方法。因而, 多个相关性状联合的混合模型方法更易被应用者接受, 其相关方法主要有GCTA[46]、MTMM[64]、GEMMA (mvLMMs)[65]、mtSet[66]和mvLMM[67], 其中GCTA只能分析2个相关性状。这些研究均表明, 多个相关性状联合分析比单个性状分析有更高的功效和精度。然而, 可供利用的Windows界面软件包还有待于研制。
5 植物关联分析的相关软件包
目前, 关联分析已在人类和动植物遗传学研究中得到广泛应用, 理论工作者也不断提出新的方法与软件(表1)。为便于应用, 这里简要介绍主要软件包。
PLINK软件[2](http://pngu.mgh.harvard.edu/~purcell/plink/)是较早开放使用的关联分析软件, 可用于数据管理、群体结构评价、复杂性状和casecontrol数据的关联分析, 也可处理基因型和表型大数据。
Cornell大学Buckler实验室开发的TASSEL软件[69](http://tassel.bitbucket.org/)是以程序设计语言Java编写的可以在主流操作系统下使用的软件包。目前已更新到TASSEL5.0版本, 主要包括关联分析、进化分析和连锁分析, 也可以计算和图示连锁不平衡统计量。2012年, 该实验室释放了基于R语言的基因关联和预测整合工具GAPIT (http://zzlab. net/GAPIT), 现已更新至GAPIT v2[70], 包含了FaST-LMM、ECMLM、FaST-LMM-Select、SUPER等关联分析新方法, 全基因组预测包含了基于CMLM、ECMLM和SUPER的gBLUP方法。新版本增加了性状表型模拟、功效分析和交叉验证等功能。
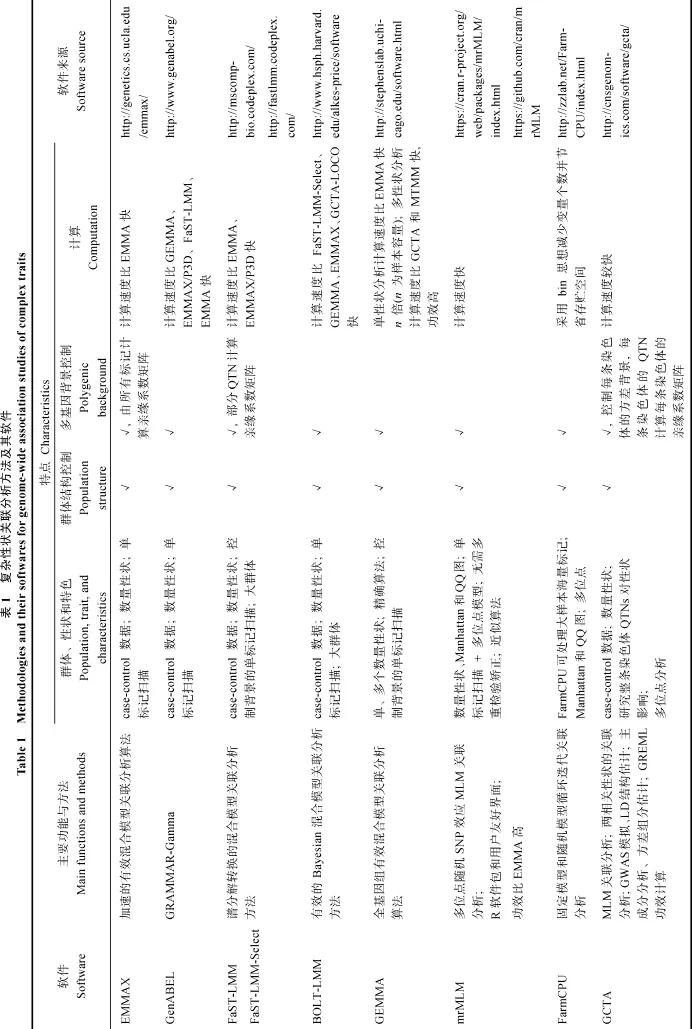

QTXNetwork是浙江大学朱军教授实验室开发的、基于GPU计算的、可以处理大规模复杂性状组学数据的关联分析软件包(http://ibi.zju.edu.cn/software/QTXNetwork/)[16,71], 包括QTL连锁分析、QTS的GWAS、QTT/P/M关联分析和GMDR全基因组关联分析数据过滤4个功能模块, 可以检测主效基因、基因与环境互作和基因间互作, 表型数据既可以是数量性状观察值又可以是组学数据, 是一款CPU与GPU异构运算平台的软件包。
mrMLM是基于Wang等[14]提出的多位点随机SNP效应混合模型方法的R软件包(https://cran.rproject.org/web/packages/mrMLM/index.html), 在R环境下可进行Windows界面操作, 在R中载入的mrMLM软件包也可在其他操作系统下运行。该软件包除多位点关联分析外, 还能提供筛选显著标记的Manhattan图和评价方法优劣的QQ (Quantile-Quantile)图。
除上述软件之外, 正文中提到的其他软件的一些相关信息可参见表1。我们相信, 新的方法与软件将不断涌现, 应用者可根据自己的需要, 选择不同的方法; 也可以用尽可能多的方法分析同一组数据,然后用逐步回归筛选出最优关联标记集。
6 展望
随着生物学组学数据、计算机科学技术和统计学算法的不断更新, 特别是植物数量性状遗传分析的需要, 有必要搭建植物关联分析的技术平台, 以剖析数量性状的遗传基础, 推动作物分子设计育种和分子生物学研究的发展(图1)。

图1 植物全基因组关联分析技术路线图Fig. 1 Technical framework for genome-wide association studies in plants
6.1海量标记高精度快速检测关联分析算法研究与软件包研制
植物关联分析方法学研究发展较快, 研究内容越来越丰富, 加快了这些方法在植物遗传研究中的应用。但是, 植物数量性状是复杂的, SNP数目远大于作图群体个体数, 使GWAS面临巨大的挑战, 特别是对于多基因检测、基因与环境互作分析[72]和基因间上位性作图。这意味着关联分析方法研究需要在统计学超饱和线性模型参数估计理论、计算机快速计算技术和矩阵论快速计算算法等方面有所突破。所以, 需要将统计方法、数值算法和计算机技术有效结合, 不断开发出新的高效、快速和海量标记的关联分析方法。为了让这些新方法得到广泛应用, 有必要研制不同平台的计算机软件包[73]。
6.2关联分析与作物育种相结合
常规的育种方法是借助表型及育种家经验对作物的重要农艺性状进行选育, 其效率低, 周期长,而基于基因型选择和高效准确的分子辅助技术, 开启了作物育种的新方向。植物重要性状关联分析的目的就是发掘有益的等位基因, 为作物育种服务。关联分析在作物育种中可快速发掘种质资源中的优异等位变异, 并通过聚合育种或其他分子设计育种方法将其引入育种材料[74-76]。但由于标记的复杂性以及遗传背景和环境的影响, 关联分析成果在分子标记辅助选择育种中的应用有待提升。此外, 通过关联分析可有助于了解目标基因的位置、遗传效应和基因网络等信息, 进而通过分子生物学操作或作物分子育种操作来改良目标性状[77]。
针对不同的育种目标可以选择不同的关联分析方法。对于纯合品种育种, 可选用上述方法; 对于杂种品种培育, 可利用育种群体进行遗传分析, 其结果可用于最优杂交组合的预测[25,58]。若要提高精度,全基因组预测是一个可供利用的方法[78]。
6.3关联分析与分子生物学和组学研究相结合
虽然关联分析能发掘更多的可供作物育种利用的等位基因, 为基因的功能分析和功能标记开发研究提供有用信息, 但是这些基因的生物学功能并不十分清楚, 只能作为植物分子生物学的前期工作。目前, 转录组、蛋白组和代谢组等组学研究十分活跃。若将这些组学数据视为复杂性状, 也可进行相应的关联分析, 在拟南芥[79]和玉米[80-81]等作物中已经得以应用。但是, 这方面的工作还需要进一步加强。
References
[1] Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science, 1996, 273: 1516–1517
[2] Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M,Bender D, Maller J, Sklar P, De Bakker P, Daly M, Sham P C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007, 81: 559–575
[3] Wan X, Yang C, Yang Q, Xue H, Fan X D, Tang N L S, Yu W C. BOOST: a fast approach to detecting gene-gene interactions in genome-wide case-control studies. Am J Hum Genet, 2010, 87: 325–340
[4] Takeuchi F, Serizawa M, Yamamoto K, Fujisawa T, Nakashima E, Ohnaka K, Ikegami H, Sugiyama T, Katsuya T, Miyagishi M,Nakashima N, Nawata H, Nakamura J, Kono S, Takayanagi R,Kato N. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes, 2009, 3: 1690–1699
[5] Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J,Lush M J, Maranian M J, Bolla M K, Wang Q, Shah M, Perkins B J, Czene K, Eriksson M, Darabi H, Brand J S, Bojesen S E, Nordestgaard B G, Flyger H, Nielsen S F, Rahman N,Turnbull C, BOCS, Fletcher O, Peto J, Gibson L, dos-Santos-Silva I, Chang-Claude J, Flesch-Janys D, Rudolph A, Eilber U,Behrens S, Nevanlinna H, Muranen T A, Aittomäki K, Blomqvist C, Khan S, Aaltonen K, Ahsan H, Kibriya M G, Whittemore A S, John E M, Malone K E, Gammon M D, Santella R M, Ursin G, Makalic E, Schmidt D F, Casey G, Hunter D J,Gapstur S M, Gaudet M M, Diver W R, Haiman C A,Schumacher F, Henderson B E, Le Marchand L, Berg C D,Chanock S J, Figueroa J, Hoover R N, Lambrechts D, Neven P, Wildiers H, van Limbergen E, Schmidt M K, Broeks A, Verhoef S, Cornelissen S, Couch F J, Olson J E, Hallberg E, Vachon C, Waisfisz Q, Meijers-Heijboer H, Adank M A, van der Luijt R B, Li J, Liu J, Humphreys K, Kang D, Choi J Y, Park S K, Yoo K Y, Matsuo K, Ito H, Iwata H, Tajima K, Guénel P,Truong T, Mulot C, Sanchez M, Burwinkel B, Marme F, Surowy H, Sohn C, Wu A H, Tseng C C, Van Den Berg D, Stram D O, González-Neira A, Benitez J, Zamora M P, Perez J I, Shu X O, Lu W, Gao Y T, Cai H, Cox A, Cross S S, Reed M W,Andrulis I L, Knight J A, Glendon G, Mulligan A M, Sawyer E J, Tomlinson I, Kerin M J, Miller N, kConFab Investigators,AOCS Group, Lindblom A, Margolin S, Teo S H, Yip C H,Taib N A, Tan G H, Hooning M J, Hollestelle A, Martens J W,Collée J M, Blot W, Signorello L B, Cai Q, Hopper J L,Southey M C, Tsimiklis H, Apicella C, Shen C Y, Hsiung C N,Wu P E, Hou M F, Kristensen V N, Nord S, Alnaes G I, NBCS,Giles G G, Milne R L, McLean C, Canzian F, Trichopoulos D,Peeters P, Lund E, Sund M, Khaw K T, Gunter M J, Palli D,Mortensen L M, Dossus L, Huerta J M, Meindl A, Schmutzler R K, Sutter C, Yang R, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Hartman M, Miao H, Chia K S,Chan C W, Fasching P A, Hein A, Beckmann M W, Haeberle L,Brenner H, Dieffenbach A K, Arndt V, Stegmaier C, Ashworth A, Orr N, Schoemaker M J, Swerdlow A J, Brinton L, Garcia-Closas M, Zheng W, Halverson S L, Shrubsole M, Long J,Goldberg M S, Labrèche F, Dumont M, Winqvist R, Pylkäs K,Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Brüning T;GENICA Network, Radice P, Peterlongo P, Manoukian S,Bernard L, Bogdanova N V, Dörk T, Mannermaa A, Kataja V,Kosma V M, Hartikainen J M, Devilee P, Tollenaar R A,Seynaeve C, Van Asperen C J, Jakubowska A, Lubinski J, Jaworska K, Huzarski T, Sangrajrang S, Gaborieau V, Brennan P,McKay J, Slager S, Toland A E, Ambrosone C B, Yannoukakos D, Kabisch M, Torres D, Neuhausen S L, Anton-Culver H,Luccarini C, Baynes C, Ahmed S, Healey C S, Tessier D C,Vincent D, Bacot F, Pita G, Alonso M R, Álvarez N, Herrero D,Simard J, Pharoah P P, Kraft P, Dunning A M,Chenevix-Trench G, Hall P, Easton D F. Genome-wide association analysis of more than 120 000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet, 2015, 47: 373–380
[6] Scuteri A, Sanna S, Chen W M, Uda M, Albai G, Strait J,Najjar S, Nagaraja R, Orrú M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret G B, Fink A A, Weder A B,Cooper R S, Galan P, Chakravarti A, Schlessinger D, Cao A,Lakatta E, Abecasis G R. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet, 2007, 3(7): e115
[7] Thornsberry J M, Goodman M M, Doebley J, Kresovich S,Nielsen D, Buckler E S. Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet, 2001, 28: 286–289
[8] Hansen M, Kraft T, Ganestam S, Säll T, Nilsson N O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet Res, 2001, 77: 61–66
[9] Zhang Y M, Mao Y C, Xie C Q, Smith H, Luo L, Xu S. Mapping quantitative trait loci using naturally occurring geneticvariance among commercial inbred lines of maize (Zea mays L.). Genetics, 2005, 169: 2267–2275
[10] Yu J, Pressoir G, Briggs W H, Vroh Bi I, Yamasaki M, Doebley J F, McMullen M D, Gaut B S, Nielsen D M, Holland J B,Kresovich S, Buckler E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet, 2006, 38: 203–208
[11] Kang H M, Zaitlen N A, Wade C M, Kirby A, Heckerman D,Daly M J, Eskin E. Efficient control of population structure in model organism association mapping. Genetics, 2008, 178: 1709–1723
[12] Zhang Z, Ersoz E, Lai C Q, Todhunter R J, Tiwari H K, Gore M A, Bradbury P J, Yu J M, Arnett D K, Ordovas J M, Buckler E S. Mixed linear model approach adapted for genome-wide association studies. Nat Genet, 2010, 42: 355–360
[13] Atwell S, Huang Y S, Vilhjálmsson B J, Willems G, Horton M,Li Y, Meng D, Platt A, Tarone A M, Hu T T, Jiang R, Muliyati N W, Zhang X, Amer M A, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker J R, Faure N, Kniskern J M,Jones J D, Michael T, Nemri A, Roux F, Salt D E, Tang C, Todesco M, Traw M B, Weigel D, Marjoram P, Borevitz J O,Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in Arabidopsis Thaliana inbred lines. J Am Soc Mass Spectrom, 2010, 465: 627–631
[14] Wang S B, Feng J Y, Ren W L, Huang B, Zhou L, Wen Y J,Zhang J, Jim M D, Xu S Z, Zhang Y M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci Rep, 2016, 6: 19444
[15] Liu X L, Huang M, Fan B, Buckler E S, Zhang Z W. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet, 2016,12(2): e1005767
[16] Zhang F T, Zhu Z H, Tong X R, Zhu Z X, Qi T, Zhu J. Mixed linear model approaches of association mapping for complex traits based on omics variants. Sci Rep, 2015, 5: 10298
[17] Devlin B, Roeder K. Genomic control for association studies. Biometrics, 1999, 55: 997–1004
[18] Song M, Hao W, Storey J D. Testing for genetic associations in arbitrarily structured populations. Nat Genet, 2015, 47: 550–556
[19] Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics, 2000, 155: 945–959
[20] Wilson L M, Whitt S R, Ibáez A M, Rocheford T R, Goodman M M, Buckler E S. Dissection of maize kernel composition and starch production by candidate gene associations. Plant Cell, 2004, 16: 2719–2733
[21] Sabatti C, Service S K, Hartikainen A L, Pouta A, Ripatti S,Brodsky J, Jones C G, Zaitlen N A, Varilo T, Kaakinen M, Sovio U, Ruokonen A, Laitinen J, Jakkula E, Coin L, Hoggart C,Collins A, Turunen H, Gabriel S, Elliot P, McCarthy M I, Daly M J, Järvelin M R, Freimer N B, Peltonen L. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet, 2009, 41: 35–46
[22] Price A L, Pattersom N J, Plenge R M, Weinblatt M E, Shadick N A, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet,2006, 38: 904–909
[23] Lee A B, Luca D, Klei L, Devlin B, Roeder K. Discovering genetic ancestry using spectral graph theory. Genet Epidemiol,2010, 34: 51–59
[24] Bulik-Sullivan B K, Loh P R, Finucane H K, Ripke S, Yang J,Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N, Daly M J, Price A L, Neale B M. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet, 2015, 47: 291–295
[25] Bu S H, Zhao X W, Yi C, Wen J, Tu J X, Zhang Y M. Interacted QTL mapping in partial NCII design provides evidences for breeding by design. PLoS One, 2015, 10(3): e0121034
[26] Li M, Liu X L, Bradbury P, Yu J M, Zhang Y M, Todhunter R J,Buckler E S, Zhang Z W. Enrichment of statistical power for genome-wide association studies. BMC Biol, 2014, 12: 73–82
[27] Kang H M, Sul J H, Service S K, Zaitlen N A, Kong S Y,Freimer N B, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet, 2010, 42: 348–354
[28] Svishcheva G R, Axenovich T I, Belonogova N M, van Duijn C M, Aulchenko Y S. Rapid variance components-based method for whole-genome association analysis. Nat Genet,2012, 44: 1166–1170
[29] Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet, 2012, 44: 821–826[30] Lippert C, Listqarten J, Liu Y, Kadie C M, Davidson R I,Heckerman D. Fast linear mixed models for genome-wide association studies. Nat Methods, 2011, 8: 833–835
[31] Listgarten J, Lippert C, Kadie C M, Davidson R I, Eskin E,Heckerman D. Improved linear mixed models for genomewide association studies. Nat Methods, 2012, 9: 525–526
[32] Loh P R, Tucker G, Bulik-Sullivan B K, Vilhjálmsson B J,Finucane H K, Salem R M, Chasman D I, Ridker P M, Neale B M, Berger B, Patterson N, Price A L. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet, 2015, 47: 284–290
[33] Wang Q, Tian F, Pan Y, Buckler E S, Zhang Z. A SUPER powerful method for genome wide association study. PLoS One, 2014, 9: e107684
[34] Zhao K, Tung C W, Eizenga G C, Wright M H, Ali M L, Price A H, Norton G J, Islam M R, Reynolds A, Mezey J, McClung A M, Bustamante C D, McCouch S R. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun, 2011, 2: 467–476
[35] Wen Z X, Tan R J, Yuan J Z, Bales C, Du W Y, Zhang S C,Chilvers M I, Schmidt C, Song Q J, Cregan P B, Wang D C. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genomics, 2014,15: 809–819
[36] Yang N, Lu Y L, Yang X H, Huang J, Zhou Y, Ali F, Wen W W,Liu J, Li J S, Yan J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet, 2014, 10(9): e1004573
[37] McCullagh P, Nelder J A. Generalized Linear Models, 2nd edn. London: Chapman and Hall, 1989
[38] Yi N, Liu N J, Zhi D G, Li J. Hierarchical generalized linear models for multiple groups of rare and common variants: jointly estimating group and individual-variant effects. PLoS Genet, 2011, 7(12): e1002382
[39] Feng J Y, Zhang J, Zhang W J, Wang S B, Han S F, Zhang Y M. An efficient hierarchical generalized linear mixed model for mapping QTL of ordinal traits in crop cultivars. PLoS One,2013, 8(4): e59541
[40] Wang L, Jia P, Wolfinger R D, Chen X, Grayson B L, Aune T M, Zhao Z. An efficient hierarchical generalized linear mixed model for pathway analysis of genome-wide association studies. BMC Bioinformatics, 2011, 27(5): 686–692
[41] Iwata H, Uga Y, Yoshioka Y, Ebana K, Hayashi T. Bayesian association mapping of multiple quantitative trait loci and its application to the analysis of genetic variation among (Oryza sativa L.) germplasms. Theor Appl Genet, 2007, 114: 1437–1449
[42] Iwata H, Ebana K, Fukuoka S, Jannink J L, Hayashi T. Bayesian multilocus association mapping on ordinal and censored traits and its application to the analysis of genetic variation among (Oryza sativa L.) germplasms. Theor Appl Genet, 2009,118: 865–880
[43] Zhang Y M, Xu S. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity, 2005, 95: 96–104
[44] Hoggart C J, Whittaker J C, De Iorio M, Balding D J. Simultaneous analysis of all SNPs in genome-wide and resequencing association studies. PLoS Genet, 2008, 4: e1000130
[45] Segura V, Vilhjálmsson B J, Platt A, Korte A, Seren Ü, Long Q,Nordborg M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet, 2012, 44: 825–830
[46] Yang J, Lee S H, Goddard M E, Visscher P M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet,2011, 88: 76–82
[47] Goddard M E, Wray N R, Verbyla K, Visscher P M. Estimating effects and making predictions from genomewide marker data. Stat Sci, 2009, 24: 517–529
[48] Zhou X, Carbonetto P, Stephens M. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet, 2013, 9(2): e1003264
[49] Moser G, Lee S H, Hayes B J, Goddard M E, Wray N R, Visscher P M. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet, 2015, 11(4): e1004969
[50] Zhang Y, Liu J S. Bayesian inference of epistatic interactions in case-control studies. Nat Genet, 2007, 39: 1167–1173
[51] Tang W W, Wu X B, Jiang R. Epistatic module detection for case-control studies: a Bayesian model with a Gibbs sampling strategy. PLoS Genet, 2009, 5(5): e1000464
[52] Cho S, Kim H, Oh S, Kim K, Park T. Elastic-net regularization approaches for genome-wide association studies of rheumatoid arthritis. BMC Proc, 2009, 3(suppl 7): S25
[53] Han B, Park M, Chen X W. A Markov blanket-based method for detecting causal SNPs in GWAS. BMC Bioinformatics, 2010, 11(suppl 3): S5
[54] Han B, Chen X W, Talebizadeh Z. FEPI-MB: identifying SNPs-disease association using a Markov blanket-based approach. BMC Bioinformatics, 2011, 12(Suppl 12): S3
[55] Li J, Dan J, Li C L, Wu R L. A model-free approach for detecting interactions in genetic association studies. Brief Bioinform, 2014, 15: 1057–1068
[56] Wang D, Eskridge K M, Crossa J. Identifying QTLs and epistasis in structured plant populations using adaptive mixed LASSO. J Agric Biol Environ Stat, 2011, 16: 170–184
[57] Lü H Y, Liu X F, Wei S P, Zhang Y M. Epistatic association mapping in homozygous crop cultivars. PLoS One, 2011, 6(3): e17773
[58] Wen J, Zhao X W, Wu G R, Xiang D, Liu Q, Bu S H, Yi C,Song Q J, Dunwell J M, Tu J X, Zhang T Z, Zhang Y M. Genetic dissection of heterosis using epistatic association mapping in a partial NCII mating design. Sci Rep, 2015, 5: 18376
[59] Aschard H, Vilhjálmsson B J, Greliche N, Morange P E,Trégouët D A, Kraft P. Maximizing the power of principalcomponent analysis of correlated phenotypes in genome-wide association studies. Am J Hum Genet, 2014, 94: 662–676
[60] Ferreira M A, Purcell S M. A multivariate test of association. Bioinformatics, 2009, 25: 132–133
[61] Bottolo L, Chadeau-Hyam M, Hastie D I, Zeller T, Liquet B,Newcombe P, Yengo L, Wild P S, Schillert A, Ziegler A, Nielsen S F, Butterworth A S, Ho W K, Castagné R, Munzel T,Tregouet D, Falchi M, Cambien F, Nordestgaard B G, Fumeron F, Tybjærg-Hansen A, Froguel P, Danesh J, Petretto E,Blankenberg S, Tiret L, Richardson S. GUESS-ing polygenic associations with multiple phenotypes using a GPU-Based evolutionary stochastic search algorithm. PLoS Genet, 2013,9(8): e1003657
[62] Bolormaa S, Pryce J E, Reverter A, Zhang Y, Barendse W,Kemper K, Tier B, Savin K, Hayes B J, Goddard M E. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet, 2014, 10: e1004198
[63] Xu Y, Hu W M, Yang Z F, Xu C W. A multivariate partial least squares approach to joint analysis for multiple correlated traits. Crop J, 2016, 4(1): 21–29
[64] Korte A, Vilhjálmsson B J, Segura V, Platt A, Long Q, Nordborg M. A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat Genet,2012, 44: 1066–1071
[65] Zhou X, Stephens M. Efficient algorithm for multivariate linear mixed models in genome-wide association studies. Nat Methods, 2014, 11: 407–409
[66] Casale F P, Rakitsch B, Lippert C, Stegle O. Efficient set tests for the genetic analysis of correlated traits. Nat Methods, 2015,12: 755–758
[67] Furlotte N A, Eskin E. Efficient multiple-trait association and estimation of genetic correlation using the matrix-variate linear mixed model. Genetics, 2015, 200: 59–68
[68] Wan X, Yang C, Yang Q, Xue H, Tang N L S, Yu W C. Predictive rule inference for epistatic interaction detection in genome-wide association studies. Bioinformatics, 2010, 26:30–37
[69] Bradbury P J, Zhang Z, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples. BMC Bioinformatics, 2007,23: 2633–2635
[70] Tang Y, Liu X, Wang J, Li M, Wang Q, Tian F, Su Z, Pan Y,Liu D, Lipka A E, Buckler E S, Zhang Z. GAPIT Version 2: Enhanced integrated tool for genomic association and prediction. Plant Genome, 2016, 9(2): doi: 10.3835/plantgenome 2015.11.0120
[71] 张福涛. 遗传分析方法的GPU并行计算与优化研究. 浙江大学博士学位论文, 浙江杭州, 2014. pp 89–97 Zhang F T. Parallelization and Optimization of GPU Computation for Genetic Analysis Methods. PhD Dissertation of Zhejiang University, Hangzhou, China, 2014. pp 89–97 (in Chinese with English abstract)
[72] Sul J H, Bilow M, Yang W Y, Kostem E, Furlotte N, He D,Eskin E. Accounting for population structure in gene-byenvironment interactions in genome-wide association studies using mixed models. PLoS Genet, 2016, 12(3): e1005849
[73] Zhang W, Dai X, Wang Q, Xu S, Zhao P X. PEPIS: a pipeline for estimating epistatic effects in quantitative trait locus mapping and genome-wide association studies. PLoS Comput Biol, 2016, 12(5): e1004925
[74] Collard B C Y, Mackill D J. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci, 2008, 363(1491): 557–572
[75] Andersen J R, Lǜbberstedt T. Functional markers in plants. Trends Plant Sci, 2003, 8: 554–560
[76] 杨小红, 严建兵, 郑艳萍, 余建明, 李建生. 植物数量性状关联分析研究进展. 作物学报, 2007, 33: 523–530 Yang X H, Yan J B, Zheng Y P, Yu J M, Li J S. Reviews of association analysis for quantitative traits in plants. Acta Agron Sin, 2007, 33: 523–530 (in Chinese)
[77] 谭贤杰, 吴子恺, 程伟东, 王天宇, 黎裕. 关联分析及其在植物遗传学研究中的应用. 植物学报, 2011, 46: 108–118 Tan X J, Wu Z K, Cheng W D, Wang T Y, Li Y. Association analysis and its application in plant genetic research. Chin Bull Bot, 2011, 46: 108–118 (in Chinese)
[78] 布素红. 多亲本群体QTL定位和优异杂交组合预测. 南京农业大学博士学位论文, 江苏南京, 2015. pp 57–68 Bu S H. Mapping of Quantitative Trait Loci and Prediction of Elite Hybrid Combination in Multi-parental Populations. PhD Dissertation of Nanjing Agricultural University, Nanjing,China, 2015. pp 57–68 (in Chinese with English abstract)
[79] Chan E K F, Rowe H C, Kliebenstein D J. Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics, 2010, 185: 991–1007
[80] Riedelsheimer C, Lisec J, Czedik-Eysenbreg A, Sulpice R, Flis A, Grieder C, Altmann T, Stitt M, Willmitzer L, Melchinger A E. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc Natl Acad Sci USA, 2012, 109: 8872–8877
[81] Wen W W, Li D, Li X, Gao Y Q, Li W Q, Li H H, Liu J, Liu H J, Chen W, Luo J, Yan J B. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun, 2014, 5: 3438–3447
Advances on Methodologies for Genome-wide Association Studies in Plants
FENG Jian-Ying1, WEN Yang-Jun1, ZHANG Jin1, and ZHANG Yuan-Ming2,*1State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, Nanjing 210095, China;2College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
Genome-wide association studies (GWAS) have been widely used in human, animal and plant genetics, and many new approaches and their softwares have been developed in recent years. To make a better use of the GWAS methods in applied research, in this study we summarized the advances on methodologies and softwares for GWAS. First, LD score regression was introduced to investigate the effect of population structure on GWAS. Then, the main approaches and their softwares for GWAS in plants were reviewed, including a single-locus model, a multi-locus model, epistasis, and multiple correlated traits. Finally, we prospected the future developments in GWAS. It should be noted that, in real data analysis at present, the methodologies for genome-wide single-marker scan under polygenic background and population structure controls are widely used,and the corresponding results are complementary to those derived from non-parameter approaches with high false discovery rate. However, the future approaches for GWAS should be based on the multi-locus genetic model, QTN-by-environment interaction, epistatic detection and multivariate analysis. Our purpose was to provide beneficial information in theoretical and applied researches.
Genome-wide association study; Epistasis; Mixed linear model; Multi-locus model
10.3724/SP.J.1006.2016.00945
本研究由国家自然科学基金项目(31301004)和中央高校基本科研业务费项目(KJQN201422)资助。
This work was supported by National Natural Science Foundation of China (31301004) and Fundamental Research Funds for the Central Universities (KJQN201422).
(Corresponding author): 章元明, E-mail: soyzhang@mail.hzau.edu.cn; Tel: 13505161564
联系方式: E-mail: fengjianying@njau.edu.cn
Received(): 2015-07-08; Accepted(接受日期): 2016-05-09; Published online(网络出版日期): 2016-05-11.
URL: http://www.cnki.net/kcms/detail/11.1809.S.20160511.1551.002.html
