特发性肺纤维化合并慢性支气管炎2例报告并文献复习
2016-09-12许霞陈会师蒲清刚王家明郑祖海
许霞,陈会师,蒲清刚,王家明,郑祖海
(阆中市人民医院呼吸内科,四川 南充 637400)
特发性肺纤维化合并慢性支气管炎2例报告并文献复习
许霞,陈会师,蒲清刚,王家明,郑祖海
(阆中市人民医院呼吸内科,四川 南充637400)
目的:提高对特发性肺纤维化合并慢性支气管炎的临床认识,探讨特发性肺纤维化发病机制。方法:回顾2例特发性肺纤维化合并慢性支气管炎的临床表现、影像学特点及诊治经过,结合有关文献进行回顾性分析。结果:2例均为老年患者,男女各1例。均在慢性支气管炎的基础上出现呼吸困难加重,胸部高分辨CT(chest high resolution,HRCT)均表现为普通型间质性肺炎特点,并排除其他已知原因的间质性肺疾病,诊断特发性肺纤维化成立。两例均接受了糖皮质激素联合N-乙酰半胱氨酸的治疗方案,随访时间1~2年,病情长期稳定,糖皮质激素均减量至10 mg/d长期维持。其中病例1最近1次住院中发现病情有所加重。结论:特发性肺纤维化患者病情呈不可逆进展,治疗无特效药物,预后不良,可酌情选用糖皮质激素联合N-乙酰半胱氨酸的方案。若合并慢性支气管炎,病情更复杂,容易导致预后不佳。
特发性肺纤维化;慢性支气管炎;糖皮质激素;N-乙酰半胱氨酸
2011年,特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)诊疗指南[1]将IPF定义为原因不明、出现在成人、局限于肺、进行性致纤维化的间质性肺炎,其组织病理学和放射学表现为普通型间质性肺炎(usual interstitial pneumonia,UIP)。该病起病隐匿、缓慢进展,最终导致呼吸衰竭,平均生存期3~5年[2],部分患者可发生急性加重,导致患者在几周或数月内死亡,病死率甚高。如果同时合并慢性支气管炎,则更增加了病情的复杂性,导致疾病诊治困难。目前国内有关IPF伴慢性支气管炎的临床报道及研究尚少。本文通过报道两例IPF伴慢性支气管炎患者,并复习相关文献,提高对该病的认识。
1 临床资料
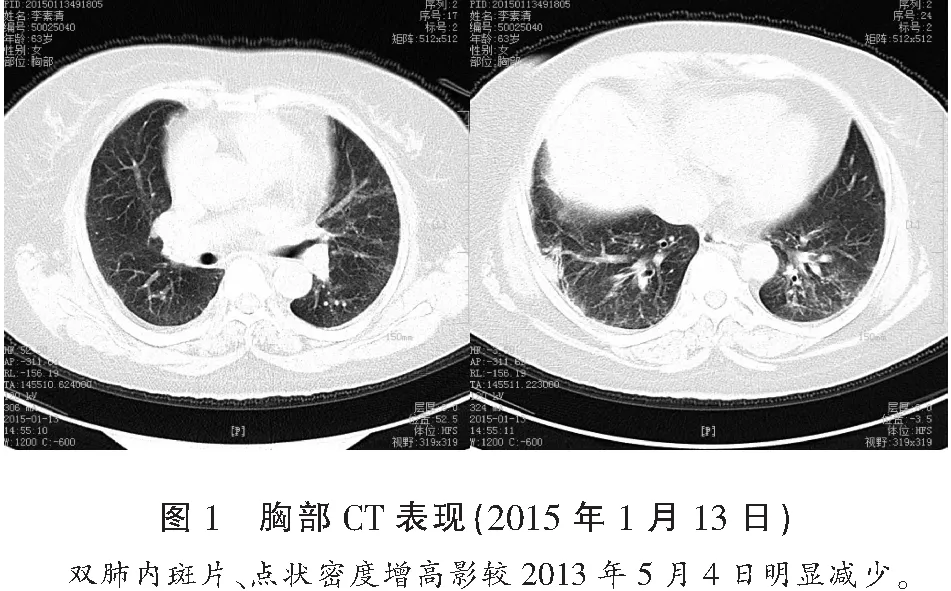
病例1:患者,女,63岁,既往有20年慢性支气管炎病史。2+年前因“咳嗽、咳痰加重6个月”在华西医院就诊,行胸部HRCT及胸腔镜下右肺切除活检,诊断为:特发性肺纤维化伴感染、慢性支气管炎,于华西医院予抗感染、祛痰、平喘等治疗,入院20 d后开始予甲强龙40 mg/d静滴,连续5 d,患者病情好转出院并遵医嘱口服强的松20 mg/d、吸入舒利迭(沙美特罗氟替卡松,50/500 μg)早晚各1喷治疗。2+月后复查胸部CT与前片比较双肺间质改变有所加重,于我院呼吸内科调整治疗方案为:强的松30 mg/d口服(按理想体重每天0.5 mg/kg计算)、舒利迭(50/500 μg)早晚各1喷吸入、富露施(N-乙酰半胱氨酸)600 mg/次(每日两次口服)等治疗,连续1个月后强的松每隔1个月减量5 mg/d至10 mg/d长期维持,长期口服富露施剂量不变,舒利迭逐渐减量,4月后因咽部不适较重停用。2015年1月,我院胸部CT示:双肺散在斑片、索条和磨玻璃样密度增高影,边界欠清,以双侧胸膜下区病变明显。双肺内斑片、点状密度增高影较2013年5月明显减少(图1),患者病情稳定。本次因“咳嗽、咳痰、喘息加重7 d”于2015年3月26日于我院呼吸内科住院。否认既往高血压病、糖尿病、冠心病病史。无吸烟、饮酒史。
入院查体:呼吸23次/min,动脉血氧饱和度93%(未吸氧)。神志清楚,呼吸急促,双肺可闻及广泛哮鸣音,双下肺可闻及较多爆裂音及中细湿罗音。
辅助检查:入院后血气分析(未吸氧):pH 7.44,动脉血氧分压 65 mmHg,二氧化碳分压36 mmHg,碳酸氢根 24.5 mmol/L。白细胞计数 9.31×109/L,中性粒细胞百分率 83.7%。痰液细菌涂片示:少量革兰氏阴性双球菌,未查见抗酸杆菌、真菌,细菌培养阴性。
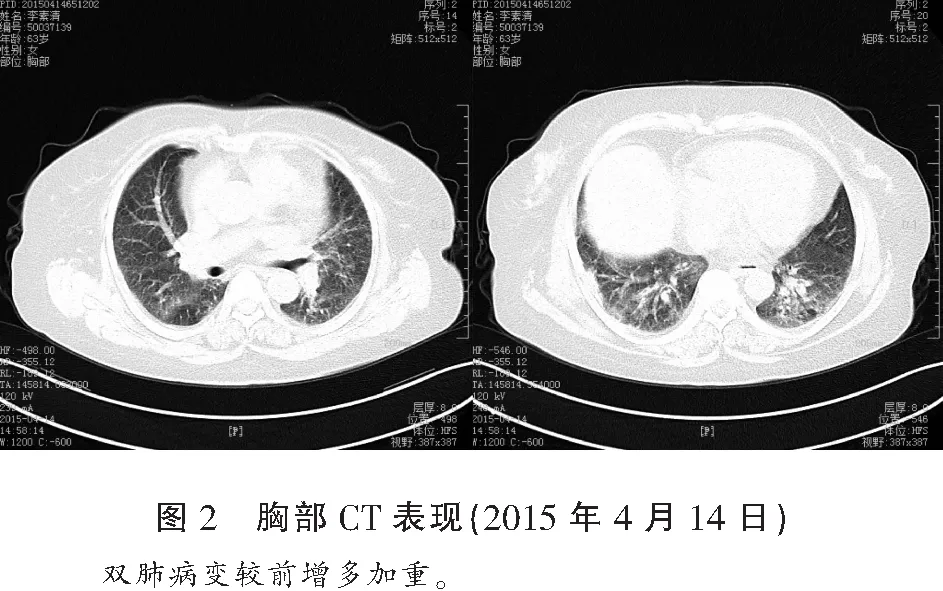
临床诊断为特发性肺纤维化伴感染、慢性支气管炎、低氧血症。先后予哌拉西林他唑巴坦、泰能、头孢美唑等静滴抗感染、祛痰、平喘、氢化可的松琥珀酸钠200 mg/8 h静滴非特异性抗炎、氧疗等治疗,静脉用激素每3~5 d减量1次,每次剂量减半或20%。经上述治疗咳嗽、咳痰及双肺哮鸣音减少,但2015年4月13日病情再次加重(住院期间),复查胸部CT示:双肺病变较前增多加重(图2)。血气分析(患者自行停止吸氧半小时):pH为7.48,动脉血氧分压为57 mmHg,二氧化碳分压为40 mmHg,碳酸氢根为29.8 mmol/L。白细胞计数 8.66×109/L,中性粒细胞百分率 78.8%。加用莫西沙星400 mg 口服 每日1次。患者病情好转于2015年4月16日出院,出院后口服甲强龙片40 mg/d(按理想体重强的松剂量每天1 mg/kg计算)连续1个月,长期口服富露施600 mg/次,每日3次。2015年5月16日患者随访病情稳定,甲强龙片减量为20 mg/d,拟该剂量维持1~2个月后酌情减量,目前仍在随访中。
病例2:患者,男,72岁,因“反复咳嗽、咳痰、喘息10+年,加重2 d”于2015年1月15日于我院呼吸内科住院。患者有10+年慢性支气管炎病史,1+年前上述症状加重,于我院呼吸内科(2013年11月11日)住院,胸部HRCT示:双肺纹理增多、增粗、紊乱交织成网格状,双肺内见广泛磨砂样、片状和网状密度增高影,密度不均,边界欠清,并散在囊状无肺纹透光区(图3)。未行活检明确,临床诊断为:特发性肺纤维化伴感染、慢性支气管炎。经治疗后好转出院,后规律予以激素治疗,此次入院2 d前受凉后咳嗽、咳痰加重,咳白色泡沫痰,量约每天40~50 mL,气促明显。既往有10+年高血压、冠心病(心绞痛型)病史,规律服用降压、冠心病预防药物,血压控制好,未再发生心绞痛。
入院查体:呼吸20次/min,动脉血氧饱和度92%(未吸氧)。双肺可闻及散在哮鸣音,双下肺可闻及较多爆裂音及中细湿罗音,双下肢无水肿。
辅助检查:入院后血气分析(未吸氧):pH 为7.42,动脉血氧分压为71 mmHg,二氧化碳分压为40 mmHg,碳酸氢根为25.9 mmol/L。白细胞计数10.01×109/L,中性粒细胞百分率为 65.3%。痰液细菌涂片示:较多革兰氏阴性双球菌,未查见抗酸杆菌、真菌,细菌培养阴性。
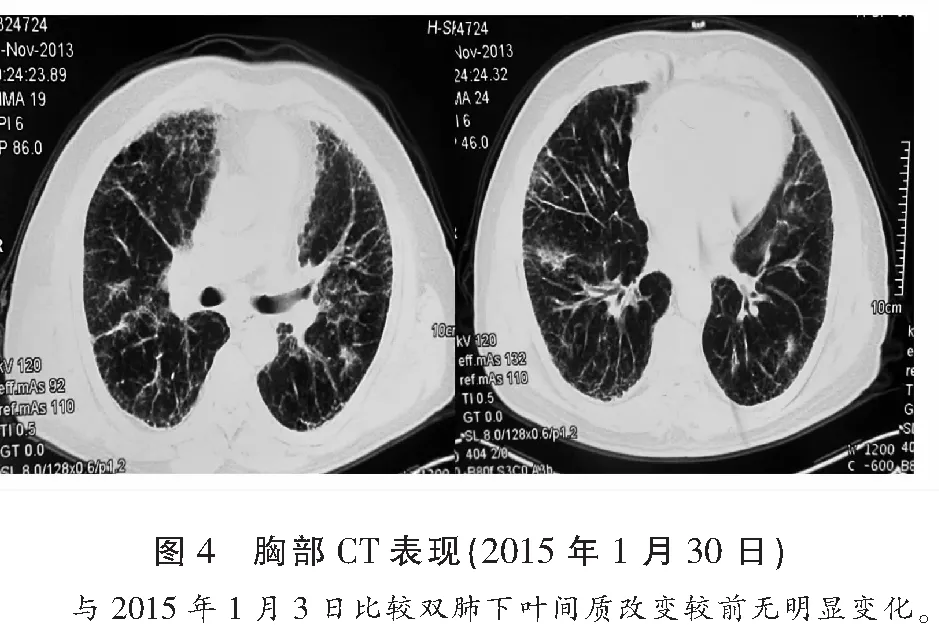
临床诊断为特发性肺纤维化伴感染、慢性支气管炎、高血压病3级,极高危组、冠心病。先后予头孢美唑联合左氧氟沙星、泰能等静滴抗感染、祛痰、平喘、氢化可的松琥珀酸钠每12小时200 mg静滴非特异性抗炎、氧疗等治疗,静脉用激素每3~5 d减量1次,每次剂量减半或20%。2015年1月30日,胸部CT与1月3日比较双肺下叶间质改变较前无明显变化(图4)。经上述治疗病情好转,咳少量白色粘痰,双肺哮鸣音消失,双下肺爆裂音减少于2015年2月10日出院,嘱出院后口服强的松25 mg/d,连续7 d,后每周减量5 mg/d至10 mg/d维持,长期口服富露施600 mg/次 每日3次,目前病情稳定,仍在随访中。
2 讨论
IPF发病率呈逐渐上升趋势,据国外调查报道,IPF在美国发生率为14~27.9例/100 000例;在欧洲为1.25~23.4例/100 000例,且发病率随年龄增长而增加[3],国内尚无相关报道。国外有研究发现IPF发病年龄多在60~70岁,男性多于女性,本文2例患者均在该年龄段发生。
IPF是一个病因复杂不明的肺部疾病,肺纤维化受到环境因素、遗传因素的影响,吸入环境因素,如空气污染、吸烟、木屑、金属粉尘、硅石、纺织灰尘、农业和牲畜都被确定为IPF发展的危险因素,其发病还与年龄、病毒和细菌感染、胃食管反流病等有关[4]。
目前国内外对于该病发病机制尚无明确定论,但大部分学者达成的共识是以肺泡上皮细胞损伤,主要是II 型上皮细胞损伤为起因引起的一系列反应,考虑可能与炎性细胞(其中以肺泡巨嗜细胞和中性粒细胞为主)、细胞因子(如TGF-β和结缔组织生长因子)、S100A9、晚期糖基化终末产物受体、转谷氨酰氨酶2等有关[5]。
IPF目前较常见的临床表现是慢性进行性呼吸困难、刺激性干咳、口唇紫绀、杵状指、肺部听诊大部分有爆裂音。诊断需排除其他已知原因的间质性肺疾病,如家庭环境、职业环境暴露、结缔组织疾病、血管炎性疾病、药物肺毒性损害等。2011年IPF诊疗指南[1]首次提出根据UIP的HRCT特点即可作为独立的IPF诊断依据。典型UIP型(符合所有4个特征):(1)病灶以胸膜下、基底部为主;(2)网格状异常改变;(3)蜂窝肺伴或不伴牵张性支气管扩张;(4)无不符合UIP型所列的特征。因国外多项研究证实HRCT诊断UIP准确性可达到90%~100%,故具有UIP典型HRCT表现者可不用行肺组织病理活检。本文2例患者HRCT均符合典型UIP特征,特发性肺纤维化诊断成立,因合并慢性支气管炎故临床表现并非刺激性干咳,而以咳痰、双肺哮鸣音为主,容易误诊为单纯的慢性阻塞性肺疾病。同时由于IPF患者肺功能检测也可能是正常的,故新指南[1]中有关IPF诊断并没有列入肺功能检查。
IPF的主要治疗药物有糖皮质激素、细胞毒药物、白三烯药物及抗纤维化药物等,2015年最新IPF国际指南中指出尼达尼布、吡啡尼酮等药物可能缓解该病进展,故推荐使用,但上述药物因价格昂贵、副作用较多,在国内尚无大量临床使用经验。目前没有任何一种药物可以逆转IPF的纤维化过程。治疗方式除了肺移植,尚无其他更有效的治疗措施。糖皮质激素被认为对大多数病例可停止症状进展或改善症状,但对于弥漫性肺纤维化疗效可能不明显[6]。目前研究发现[7]糖皮质激素类药物仅对20%的IPF患者有效,并且长期大量服用极易引发严重的不良反应,因此目前已不推荐单独使用糖皮质激素用于IPF的维持治疗,仅其在IPF急性加重时可短期使用。N-乙酰半胱氨酸是氧自由基清除药谷胱甘肽的前体,付晓巍等[8]报道长期使用该药抗氧化治疗可能改善IPF患者的临床情况及肺功能,
且不良反应小,安全性高,价格较便宜,在IPF患者知情同意和有强烈药物治疗愿望时,推荐选择该药。国外已有一项治疗性研究[9]发现激素+硫唑嘌呤+乙酰半胱氨酸联合治疗组在进行至中期32周时,联合治疗组明显增加死亡和住院的风险,主要终点指标FVC联合用药组下降0.24 L,而安慰剂组FVC联合用药组下降0.23 L,现已终止试验,结论是不支持联合治疗,故目前三药联合使用仅被推荐用于小范围的IPF患者。对肺纤维化有一定治疗作用的中药复方包括抗纤汤、复方鳖甲、益阴活血汤、肺纤康、补气通肺饮、乌蛇散等,具有毒性低、不良反应少等特点,为中药治疗肺纤维化带来了可喜的应用前景[10]。本文两例患者均采用糖皮质激素联合N-乙酰半胱氨酸的方案,随访时间在1~2年,病情长期稳定,糖皮质激素均减量至10 mg/d长期维持,治疗有效,故糖皮质激素联合N-乙酰半胱氨酸的方案可酌情使用,目前对于糖皮质激素起始剂量、如何减量维持尚无统一标准,应重视个体化,尽量减少激素的全身副作用。该研究由于病例数偏少,有待进一步研究,但为该病的治疗提供了新的治疗思路。病例1最近1次住院中发现病情有所加重,考虑与肺纤维化进展、感染等综合因素有关。本文两例患者均为IPF合并慢性支气管炎由于双重的病理损伤,使肺组织受到严重破坏,造成肺的通气与换气功能严重障碍,势必将影响患者预后[11]。
IPF的发病机制尚不明确,病情呈不可逆进展,治疗无特效药物,预后不良。该病的诊治研究仍然任重道远,临床工作者需要不断提高对该病的认识水平,积极开展多学科研究讨论,从而制定有效的治疗措施,减少和预防急性加重的发生、改善患者生活质量及生存率。
[1]Raghu G,Collard HR,Egan JJ,et al.An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis:evidence-based guidelines for diagnosis and management [J].Am J Respir Crit Care Med,2011,183(6):788-824.
[2]Ambrosini V,Cancellieri A,Chilosi M,et al.Acute exacer-bation of idiopathic pulmonary fibrosis:report of a series[J].Eur Respir J,2003,22(5):821-826.
[3]Nalysnyk L,Cid-Ruzafa J,Rotella P,et al.Incidence and prevalence of idiopathic pulmonary fibrosis:review of the literature[J].Eur Respir Rev,2012,21(126):355-361.
[4]王小华,段军.特发性肺纤维化病因学的新进展[J].医学综述,2014,20(3):414-416.
[5]赵静,刘瑞娟.特发性肺间质纤维化的相关研究进展[J].山东医药,2014,54(7):88-90.
[6]朱玉龙,张建.特发性肺间质纤维化76例临床分析[J].临床肺科杂志,2009,14(10):1370-1371.
[7]潘有禄,黄文海,沈正荣,等.肺纤维化发生机制及治疗研究进展[J].中国药学杂志,2012,47(23):1873-1876.
[8]付晓巍,童翔,范红,等.乙酰半胱氨酸治疗特发性肺纤维化的Meta分析[J].中国询证医学杂志,2014,14(4):449-455.
[9]IPF Clinical Research Network,Raghu G,Anstrom KJ,et al.Prednisone,azathioprine,and N-Acetylcysteine for pulmonary fibrosis[J].N Engl J Med,2012,366(21):1968-1977.
[10]刘芳,赵建,丁日高,等.特发性肺纤维化防治研究进展[J].人民军医,2014,57(4):451-453.
[11]王玲,居来提,李凤森,等.慢性阻塞性肺疾病合并肺间质纤维化预后观察[J].中国现代医生,2011,49(5):141,146.
(学术编辑:陈绍平)
2 cases reports and literature review of idiopathic pulmonary fibrosis combined with chronic bronchitis
XU Xia,CHEN Hui-shi,PU Qing-gang,WANG Jia-ming,ZHENG Zu-hai
(Department of Respiratory Medicine,People’s Hospital of Langzhong,Langzhong 637400,Sichuan,China)
【Abstract】Objective:To improve the clinical understanding of idiopathic pulmonary fibrosis(IPF)combined with chronic bronchitis and investigate pathogenesis of IPF.Methods:To review the clinical manifestations,imaging features and diagnosis and treatment of 2 cases of IPF combined with chronic bronchitis,and review the relevant literatures.Results:Two patients were more than 60 years old.One was female,and the other male.They had a history of chronic bronchitis,and felt aggravated dyspnea.Chest high resolution CT (HRCT)showed the feature of usual interstitial pneumonia,and other known causes of interstitial lung disease were excluded.So diagnosis of IPF was established.Two patients received the treatment of glucocorticoid combined with N-cysteine,the follow-up period was 1+to 2+years with the long-term stability of the disease,and the maintenance dose of glucocorticoids is 10 mg/d.In addition the first patient was found to be aggravated during the last hospitalization.Conclusion:The progression of IPF was irreversible and the prognosis was not good.The disease has no specific treatment drugs.We can choose the treatment of glucocorticoid combined with N-cysteine.If IPF was combined with chronic bronchitis,the disease is more complex and it will lead to poor prognosis of patients.
Idiopathic pulmonary fibrosis;Chronic bronchitis;Glucocorticoid;N-cysteine
10.3969/j.issn.1005-3697.2016.04.043短篇论著
2016-02-28
许霞(1979-),女,主治医师,硕士研究生。E-mail:185746572@qq.com
网络出版时间:2016-8-217∶48网络出版地址:http://www.cnki.net/kcms/detail/51.1254.R.20160802.1748.086.html
1005-3697(2016)04-0602-04
R734.2;R563
A
