甘草查尔酮A抑制ERK1/2/NF-κB途径减轻吸烟诱导的小鼠急性肺损伤
2016-09-10任倩倩汪丽佩谢强敏张水娟浙江中医药大学药学院浙江杭州005浙江中医药大学基础医学院浙江杭州005浙江中医药大学附属第一医院浙江杭州0006浙江大学医学院呼吸药物研究实验室浙江杭州0058
任倩倩,汪丽佩,赵 炜,陆 红,谢强敏,张水娟(.浙江中医药大学药学院,浙江杭州 005;.浙江中医药大学基础医学院,浙江杭州 005;.浙江中医药大学附属第一医院,浙江杭州0006;.浙江大学医学院呼吸药物研究实验室,浙江杭州 0058)
甘草查尔酮A抑制ERK1/2/NF-κB途径减轻吸烟诱导的小鼠急性肺损伤
任倩倩1,汪丽佩2,赵炜3,陆红1,谢强敏4,张水娟1
(1.浙江中医药大学药学院,浙江杭州310053;2.浙江中医药大学基础医学院,浙江杭州310053;
3.浙江中医药大学附属第一医院,浙江杭州310006;4.浙江大学医学院呼吸药物研究实验室,浙江杭州310058)
网络出版时间:2016-4-26 11:06网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20160426.1106.022.html
目的探讨甘草查尔酮A(LA)对吸烟诱导的急性肺损伤小鼠的保护作用及相关机制。方法整体实验:香烟烟雾暴露构建小鼠急性肺损伤模型,取肺泡灌洗液(BALF)进行白细胞计数,测定肺内角质形成细胞衍生趋化因子(KC)、肿瘤坏死因子α(TNF-α)、白介素-1β(IL-1β)和基质金属蛋白酶9(MMP-9)的mRNA和蛋白表达水平,用试剂盒测定肺组织中过氧化物髓化酶(MPO)、超氧化物歧化酶(SOD)活力以及谷胱甘肽(GSH)水平。肺组织病理切片进行HE染色。离体实验:香烟烟雾提取物(CSE)诱导上皮细胞损伤,测定细胞内白介素-8(IL-8)、MMP-9 mRNA的表达。Western blot分析细胞外信号调节激酶l/2(ERK1/2)、p38分裂原活化蛋白激酶(p38 MAPK)、c-Jun氨基末端激酶(JNK)的磷酸化水平和转录因子NF-κB p65的活性。结果整体实验显示LA组肺内炎症细胞数量及浸润程度均明显低于模型组。模型组肺内KC、TNF-α、IL-1β、MMP-9 mRNA和蛋白表达较正常对照组均明显升高,经LA处理后,上述指标相比模型组明显降低。与正常对照组相比,模型组肺内MPO活性明显上升,而SOD活性和GSH水平明显下降。LA能逆转这种变化,明显降低MPO活性,升高SOD活性和GSH水平。离体实验显示,LA(2.5、5 μmol·L-1)能明显降低CSE刺激的细胞内IL-8和MMP-9 mRNA的高表达。CSE可以诱导细胞ERK1/2磷酸化和胞核内NF-κB p65的表达,经LA预处理后,上述指标可明显抑制。结论LA可能通过阻断ERK1/ 2/NF-κB途径来抑制炎症介质的高表达、调节氧化/抗氧化失衡、下调金属蛋白酶,从而发挥对急性肺损伤小鼠的保护作用。
甘草;甘草查尔酮A;肺损伤;吸烟;肺上皮细胞;细胞因子;信号通路
慢性阻塞性肺部疾病(chronic obstructive pulmonary diseases,COPD)是一种以气流受限为特征的疾病,它与肺部对有害气体或有毒颗粒的异常炎症反应有关[1]。COPD目前已成为影响人类生活质量的全球性健康问题,发病率呈现逐年增加的趋势,迄今尚无药物能遏制COPD病情的进行性发展,因此迫切需要发展新的COPD治疗方法。从中药和植物药中寻找新的有效成分,或从已有的有效成分中发现新的药理作用是一个重要方向。尽管COPD发病机制尚未完全阐明,但目前普遍认为,吸烟是COPD发生的最主要因素之一[2],吸烟能诱导炎症并直接导致肺组织的损伤[3]。COPD的病理生理除小气道的炎症外,氧化与抗氧化失衡也是一个很重要的因素[4]。因此,从中药中寻找抗炎和抗氧化的治疗COPD的新药是一个值得探索的途径。甘草是中药中传统的镇咳祛痰药,入药历史悠久,性味甘平,归脾、胃、心、肺经,具有益气补中、润肺止咳、清热解毒、缓急止痛、调和诸药等功效。甘草中主要含有甘草酸、甘草次酸、黄酮、生物碱和氨基酸等活性成分[5]。甘草中提取的黄酮单体甘草查尔酮 A (LA)经研究表明具有抗炎、抗氧化和抗肿瘤等作用,能通过调节丝裂原活化蛋白激酶(MAPK)信号通路,诱导人胃癌细胞的凋亡[6-8]。目前LA对肺损伤的研究也有报道,研究发现其能抑制细菌内毒素脂多糖(LPS)气道滴入引起的小鼠急性肺损伤,降低肺内肿瘤坏死因子α(TNF-α)、白介素-1β(IL-1β)和白介素-6(IL-6)表达。进一步的研究发现,LA主要是通过NF-κB和p38/ERK MAPK信号途径起作用[9-10],但LA与香烟烟雾所引起的疾病之间的关系研究,无论是整体上还是离体上均未见到相关的报道。为此,本研究采用香烟烟雾诱导建立小鼠急性肺损伤模型,同时用香烟烟雾提取物(CSE)诱导肺上皮细胞损伤,观察LA对肺损伤病理改变、炎症因子、氧化/抗氧化水平、MAPK信号途径和胞核内NF-κB p65表达的影响,探索LA对香烟烟雾诱导的小鼠急性肺损伤的保护作用及其相关机制。
1 材料与方法
1.1实验动物与药品C57BL/6清洁级小鼠,♀,体质量(20±2)g,由上海斯莱克实验动物有限责任公司提供,实验动物质量合格证:SCXK(沪)2012-0002,饲养于浙江大学动物实验中心SPF级动物房。人肺上皮细胞BEAS-2B细胞株,购自美国菌种保藏中心(ATCC)细胞库。LA委托沈阳药科大学中药学院赵余庆教授课题组提取和纯化,纯度>98%。3R4F标准香烟购自美国肯塔基大学烟草研究所(批号200712)。RT-PCR相关试剂购自日本TaKaRa公司。ELISA试剂盒购于eBioscience公司。过氧化物髓化酶(MPO)、超氧化物歧化酶(SOD)以及谷胱甘肽(GSH)测定试剂盒购自南京建成生物技术有限公司。ERK1/2、p-ERK1/2、p-p38、p38、JNK、p-JNK、NF-κB p65抗体购于Cell Signaling公司。
1.2模型制备与分组给药整体实验中,将60只♀C57BL/6清洁级小鼠随机分为正常对照组、模型组、LA低、中、高剂量组(10、20、40 mg·kg-1)、地塞米松对照组(Dex,1 mg·kg-1)。依据我们以前报道的方法,建立香烟烟雾诱导的小鼠急性肺损伤模型[11]。香烟烟雾连续暴露4 d,正常对照组使用空气进行暴露。各组小鼠预给药2 d,每天给药1次,而后在连续4 d的烟雾暴露前1 h灌胃给予药物。末次香烟烟雾暴露结束后18h处理动物。
在离体实验中,依据我们以前报道的方法制作CSE,用CSE刺激人肺上皮细胞(BEAS-2B),诱导上皮细胞损伤[12]。采用不同浓度的 LA(1、2.5、5 μmol·L-1)预处理1 h,而后加入2.5%CSE诱导上皮细胞损伤,48 h后提取细胞RNA进行细胞因子测定;2.5%CSE诱导30min后提取细胞蛋白进行信号通路研究。
1.3小鼠肺泡灌洗液(BALF)制备和炎症细胞计数末次香烟烟雾暴露结束后18 h,腹腔注射6 g· kg-1乌拉坦处死各组小鼠。分离小鼠气道,插管,结扎左肺,进行肺泡灌洗,取50 μL肺泡灌洗液,加同体积白细胞计数液,100倍光镜下进行白细胞计数;其余肺泡灌洗液4℃ 2 000 r·min-1离心10min,沉淀做细胞涂片,室温干燥后瑞氏-吉姆萨染色,400倍光镜下依据形态学标准进行细胞分类计数。
1.4肺组织病理学观察按照我们以前报道的方法[11-12],将肺组织福尔马林固定,石蜡包埋,切片,HE染色,200倍镜下检查肺组织炎症细胞浸润程度。
1.5ELISA取小鼠肺泡灌洗液,根据ELISA试剂盒说明书,分别检测灌洗液中角质形成细胞衍生趋化因子(KC)、TNF-α、IL-1β和基质金属蛋白酶9 (MMP-9)蛋白含量。
1.6MPO、SOD和GSH水平测定取肺组织,制成5%肺匀浆,按试剂盒说明书分别测定上清液中MPO、SOD活力以及GSH的水平。
1.7荧光实时定量PCR测定KC、TNF-α、IL-1β、MMP-9和白介素-8(IL-8)mRNA表达水平用TRIzol试剂提取小鼠肺组织及细胞样本中总RNA,参照逆转录试剂盒说明书合成cDNA,引物序列由上海生工生物技术有限公司合成,详见Tab 1。用cDNA模板对相关基因分别进行PCR扩增,扩增条件:95℃ 预变性2 min,95℃变性10 s,退火58℃10 s,72℃延伸30 s,共40个循环。以β-actin为内参照,采用ΔΔCt值法测定基因表达水平。

Tab 1 Primer sequences used in the present study
1.8Western blot将处于对数生长期的BEAS-2B细胞接种至6孔板中,相应处理后,弃培养液,提取细胞蛋白。制备SDS-PAGE凝胶,上样、电泳、转膜,室温下用5%脱脂奶粉封闭2 h,取出硝酸纤维素膜,放入兔抗ERK1/2、p-ERK1/2、p-p38、p38、JNK、p-JNK、NF-κB p65一抗液中,4℃过夜。次日洗膜后加入荧光二抗,室温避光摇动2 h,用Odyssey荧光扫描仪,拍摄条带,采用Quantity one软件计算灰度值。
1.9统计学处理采用SPSS(20.0版)统计软件包进行数据分析,实验数据以±s表示,采用单因素方差分析(one-way ANOVA)比较多组均数以及SNK检验进行两两之间的比较。
2 结果
2.1LA对肺部炎症细胞的影响对吸烟小鼠肺泡灌洗液中炎症细胞进行总数和分类计数,如Fig 1所示,与正常对照组小鼠相比,模型组小鼠BALF中白细胞总数、中性粒细胞、淋巴细胞和巨噬细胞数均有明显增加(P<0.01)。LA能明显抑制吸烟诱导小鼠BALF中白细胞总数、中性粒细胞的增加,呈明显的量效关系。LA 40 mg·kg-1组对淋巴细胞和巨噬细胞的增加亦有明显抑制作用。小鼠肺组织HE染色后发现,香烟烟雾暴露的小鼠肺组织中性粒细胞和巨噬细胞浸润增加,而LA能明显抑制吸烟小鼠中性粒细胞和巨噬细胞在肺部的浸润,见Fig 2。
2.2LA对肺内细胞因子的影响为了探讨LA是否通过抑制炎症反应而保护小鼠急性肺损伤,应用荧光实时定量PCR检测各组小鼠肺组织细胞因子mRNA表达及ELISA法检测BALF中蛋白水平。结果显示,与正常对照组相比,模型组小鼠肺组织中KC、TNF-α、IL-1β和 MMP-9 mRNA的表达(Fig 3)及BALF中蛋白含量(Fig 4)均明显升高。与模型组相比,LA 20、40 mg·kg-1组上述指标均明显降低。
2.3LA的抗氧化效应为了探讨LA是否通过调节氧化应激反应而保护吸烟诱导的急性肺损伤小鼠,用试剂盒测定各组小鼠肺组织中MPO、SOD和GSH的水平。结果显示,与正常对照组相比,模型组小鼠肺组织中MPO水平明显上升(P<0.01),SOD和GSH水平明显下降(P<0.05~0.01)。LA能逆转这种变化,明显降低MPO水平,升高SOD和GSH水平,见Fig 5。 Fig 1LA inhibits inflammatory cell accumulation in BALF of cigarette smoke-exposed mice(±s,n=10)
A:Total leukocyte cell counts;B:Differential cell counts.##P<0.01 vs control group;*P<0.05,**P<0.01 vs model group.Licochalcone A(LA);Dexamethasone(Dex)

Fig 2 LA inhibits inflammatory cells’infiltration in lungs of cigarette smoke-exposed mice(HE,×200)

Fig 3 LA inhibits expression of cytokines mRNA in lung tissues of cigarette smoke-exposed mice(±s,n=10)
2.4LA抑制CSE刺激BEAS-2B细胞表达IL-8 和MMP-9 mRNA利用荧光实时定量PCR检测各组细胞中IL-8和MMP-9 mRNA的表达。结果显示,与空白对照组相比,2.5%CSE刺激细胞48 h后,IL-8和MMP-9 mRNA的表达明显上升(P<0.01),而2.5、5 μmol·L-1的LA能明显抑制两者的上升(P<0.05~0.01)。不加CSE刺激,只单独给予5 μmol·L-1LA的细胞,IL-8和MMP-9 mRNA的表达与空白对照组细胞(不加CSE,不加药物)相当,无明显变化,见Fig 6。
2.5LA对CSE刺激BEAS-2B细胞信号通路活化的影响通过Western blot方法检测BEAS-2B细胞p38、JNK和ERK1/2磷酸化表达,探索LA治疗小鼠急性肺损伤的作用机制。如Fig 7所示,CSE可以诱导p38、JNK和ERK1/2的磷酸化,而LA能剂量依赖性抑制由CSE诱导的肺上皮细胞ERK1/2磷酸化(Fig 7C),但对p38(Fig 7A)和JNK(Fig 7B)磷酸化无明显抑制作用。采用核质分离的方法检测细胞核内NF-κB亚基p65的表达,以进一步探索LA治疗小鼠急性肺损伤的作用机制。如Fig 7D所示,2.5%CSE能明显诱导p65的表达,LA可明显抑制其表达。
3 讨论
COPD以遍及气道、肺实质和肺血管的慢性炎症为特征。吸烟是诱发COPD的主要因素之一,香烟烟雾中含有数以万计的有毒有害物质,这些有毒化合物一旦进入肺部,就难以代谢排出,从而诱导炎症并直接导致肺组织的损伤[13]。本研究重点观察LA对吸烟诱导的小鼠急性肺损伤的作用及作用机制。
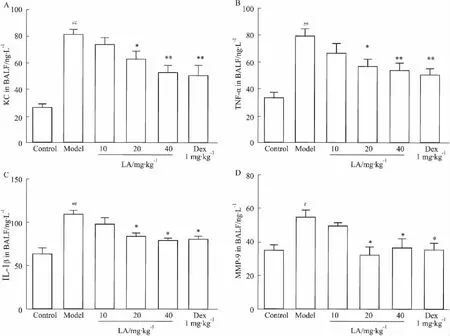
Fig 4 LA inhibits expression of cytokines protein in BALF of cigarette smoke-exposed mice(±s,n=10)
COPD的气道炎症不同于哮喘,其炎症细胞主要为中性粒细胞、CD8+T淋巴细胞和巨噬细胞,肺损伤后炎症细胞的浸润、活化导致促炎细胞因子的异常表达,与COPD的病理生理过程密切相关。KC是人IL-8的同源物,通过其受体CXCR2产生作用,是肺中性粒细胞趋化的重要介质。有研究显示,通过小分子抑制剂抑制CXCR2受体,能明显抑制吸烟诱导动物模型中肺中性粒细胞炎症[14]。前炎细胞因子,如TNF-α和IL-1β,能增强炎症反应。吸烟可介导COPD患者全身性炎症反应,与非吸烟者相比,吸烟的COPD患者血清中TNF-α表达明显升高[15]。最近,IL-1β因其参与炎症反应的长期持续过程而广受关注[16]。IL-1β能明显激活COPD患者体内的巨噬细胞,释放炎症细胞因子、趋化因子和MMP-9[17]。本研究中,香烟暴露4 d后,小鼠肺部炎症细胞(中性粒细胞和巨噬细胞)大量聚集,肺泡灌洗液和肺组织中炎症因子KC、TNF-α及IL-1β的蛋白和mRNA水平明显上升。LA能有效抑制这些炎症细胞的聚集和炎症因子的释放,提示其对肺损伤的保护作用可能与抑制炎症反应有关。
体内氧化/抗氧化平衡失调是COPD慢性损伤的重要原因。人们发现,无论在急性发作期还是在缓解期均存在两者作用失调。烟草中含有大量的氧化剂,刺激炎症细胞生成内源性氧化物,减少抗氧化物生成,促进氧化应激反应。SOD和GSH是气道内的抗氧化物质,研究表明香烟烟雾可以降低内源性抗氧化物的活性。如在人支气管肺泡上皮细胞实验中,发现香烟提取物能降低GSH的含量[18],而另一项临床研究发现,吸烟者血浆中 SOD的活性下降[19]。研究显示,LA能抑制由叔丁基过氧化氢诱导的氧化应激反应,降低氧基自由基离子(ROS)水平,减少GSH的消耗[20]。我们的研究发现,LA不仅能够抑制炎症因子的释放,而且能够有效降低MPO活性,同时升高SOD活性和GSH水平。我们的结果表明,在香烟诱导的小鼠模型中,LA对肺损伤的保护作用可能与调节氧化和抗氧化的平衡有关。
正常人体的气道、肺泡内蛋白酶和抗蛋白酶处于平衡状态,这种平衡的失调,会导致肺实质的结构破坏和纤维化。目前认为肺泡损伤和小气道纤维化是COPD气流受限的重要原因[21]。基质金属蛋白酶(matrix metalloproteinases,MMPs)是一组锌离子依赖性蛋白水解酶家族,能降解细胞外基质蛋白。MMP-9是该家族最重要的成员之一,在纤维化修复过程中,它是参与细胞外基质成分降解的关键酶[22]。研究显示,在香烟烟雾诱导的小鼠肺部炎症模型中,MMP-9表达明显上升[23],另有研究发现COPD患者的MMP-9表达较正常人高[24]。本实验结果显示,香烟暴露后,小鼠肺泡灌洗液内MMP-9蛋白表达增加,而LA能够明显抑制这种蛋白的表达。MMP-9 mRNA的表达变化和其蛋白一致,在整体和离体实验中,LA均能明显抑制MMP-9 mRNA的高表达。我们推测LA对香烟诱导的急性肺损伤的保护作用可能与下调蛋白酶有关。

Fig 5 LA inhibits oxidative stress in lung tissues of cigarette smoke-exposed mice(±s,n=10)
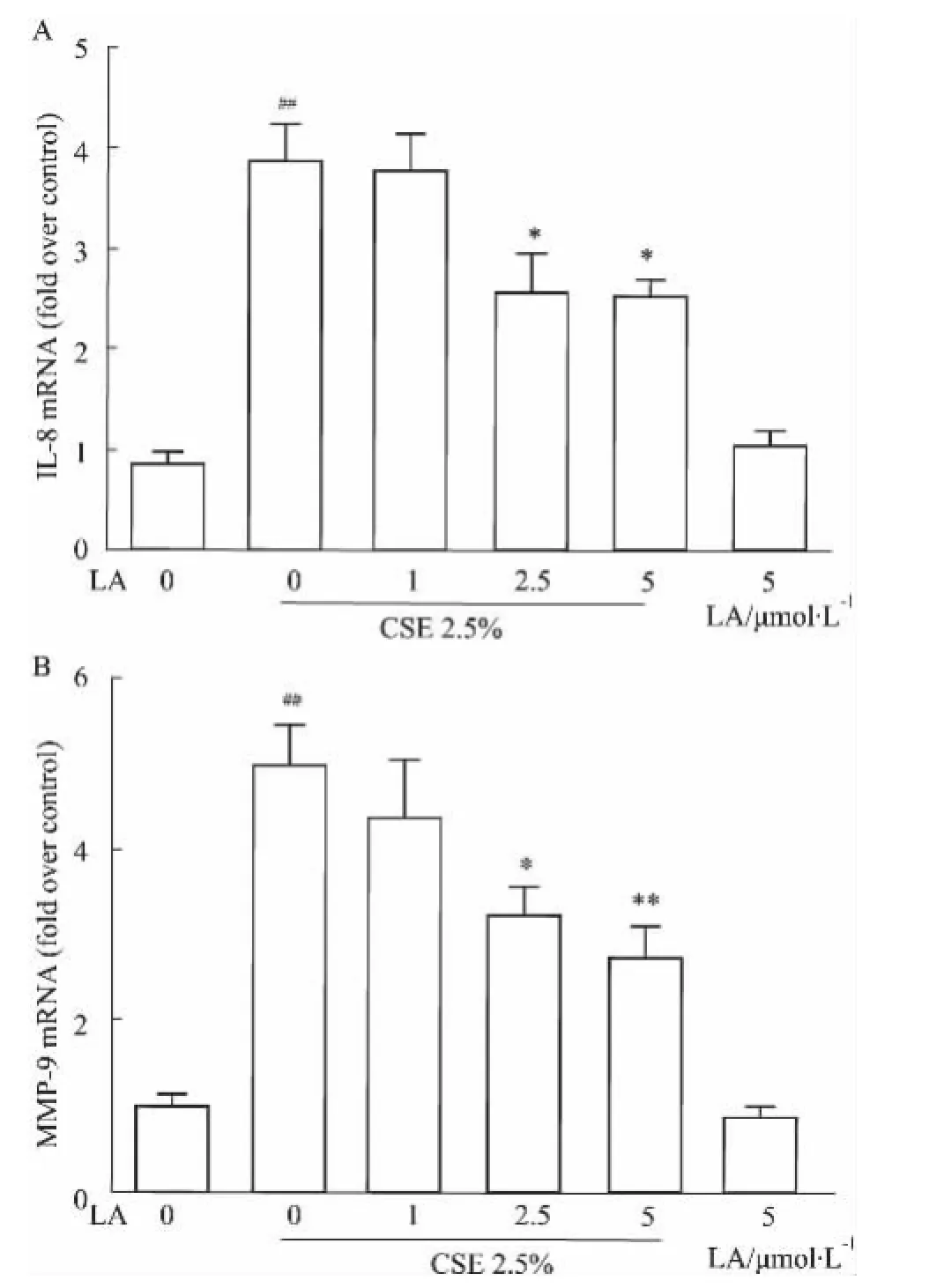
?Fig 6 LA suppresses CSE-induced mRNA expression of cytokines in BEAS-2B cells(±s,n=6)
肺上皮屏障是肺抵抗外界环境的第一道防御系统,在这个屏障里,肺上皮细胞起很重要的防御作用。为进一步探索LA对急性肺损伤的保护作用和作用机制,我们用人肺上皮细胞株BEAS-2B进行了相关研究。目前的研究[25]和我们的实验均显示,在香烟烟雾刺激的人肺上皮细胞中,IL-8表达量的升高幅度比其他炎症因子高。炎症细胞的迁移和激活受到细胞因子和趋化因子的调节,IL-8又称CXCL8,是中性粒细胞趋化因子,在COPD发展过程中发挥重要作用[26]。Schneider等[27]的研究显示,COPD患者气道上皮细胞中 IL-8表达明显升高。IL-8通过其受体CXCR2发挥作用,用CXCR2拮抗剂可明显减轻中性粒细胞炎症和肺泡损伤。本研究中,2.5%CSE刺激细胞48 h后,IL-8 mRNA的表达明显上升,而LA能明显抑制其高表达,进一步证明了LA对肺损伤中炎症反应的抑制作用。

Fig 7 Effects of LA on CSE-induced protein expression of MAP kinases and NF-κB in BEAS-2B cells(±s,n=6)
MAPK家族包括ERK(extracellular signal-regulated kinase)、p38和JNK(c-Jun NH2-terminal kinase),这些通路调节细胞的多种生理病理过程,如细胞增殖、分化、迁移、存活和死亡等。转录因子NF-κB是内皮激活过程中重要的调节因子,与许多炎症因子的表达相关,包括细胞因子和黏附分子NO、TNF-α、MCP-1和CAMs[28-29]。在细胞核内,NF-κB以p65/ p50二聚体形式存在,NF-κB的激活过程首先是与IκBα解离,然后p65亚基入核,介导炎症因子的转录活性[30]。研究报道,CSE诱导的炎症反应与MAPK信号通路的激活密切相关,阻断MAPK通路的激活能保护上皮细胞,减轻炎症反应、抗凋亡和抗氧化应激反应[31-32]。Chu等[9]的研究证明LA能通过p38/ERK MAPK信号通路缓解LPS诱导的小鼠急性肺损伤模型的炎症反应,Furusawa等[10]还发现其能明显抑制LPS诱导的NF-κB转录活性。我们猜测LA调节CSE诱导的炎症反应可能与MAPK途径中的多条信号通路及NF-κB的核转录有关。为了验证这一推测,我们采用Western blot方法检测BEAS-2B细胞中p38、JNK和ERK1/2的磷酸化表达以及NF-κB的活性。实验结果表明,LA能明显抑制CSE诱导的ERK1/2的磷酸化,但对p38和JNK的磷酸化无明显作用,同时LA也能有效阻断细胞核内NF-κB p65亚基的激活,提示LA发挥的抗炎作用可能是通过阻断NF-κB p65的激活以及抑制CSE诱导的ERK1/2 MAPK的激活。
综上所述,LA对香烟烟雾诱导的急性肺损伤具有保护作用,这种保护作用可能与抑制炎症介质的高表达、调节氧化/抗氧化失衡、下调金属蛋白酶有关,其调控机制可能与阻断ERK1/2/NF-κB途径有关。
(致谢:本实验在浙江大学医学院呼吸药物研究实验室完成。感谢谢强敏教授和实验室同学在本课题研究中给予的指导和帮助。感谢沈阳药科大学中药学院赵余庆教授课题组提取和纯化甘草查尔酮A。)
[1]Vestbo J,Hurd S S,Agustí A G,et al.Global strategy for the diagnosis,management,and prevention of chronic obstructive pulmonary disease:GOLD executive summary[J].Am J Respir Crit Care Med,2013,187(4):347-65.
[2]Barreiro E,Peinado V I,Galdiz J B,et al.Cigarette smoke-induced oxidative stress:a role in chronic obstructive pulmonary disease skeletal muscle dysfunction[J].Am J Respir Crit Care Med,2010,182(4):477-88.
[3]Crotty Alexander L E,Shin S,Hwang J H.Inflammatory diseases of the lung induced by conventional cigarette smoke:a review [J].Chest,2015,148(5):1307-22.
[4]Matera M G,Calzetta L,Cazzola M.Oxidation pathway and exacerbations in COPD:the role of NAC[J].Expert Rev Respir Med,2016,10(1):89-97.
[5]Zhang Q,Ye M.Chemical analysis of the Chinese herbal medicine Gan-Cao(licorice)[J].J Chromatogr A,2009,1216(11):1954-69.
[6]Kühnl J,Roggenkamp D,Gehrke S A,et al.Licochalcone A activates Nrf2 in vitro and contributes to licorice extract-induced lowered cutaneous oxidative stress in vivo[J].Exp Dermatol,2015,24(1):42-7.
[7]Hao W,Yuan X,Yu L,et al.Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways[J].Sci Rep,2015,5:10336.
[8]王艳明,刘瑛,阎新燕,等.甘草查尔酮A抑制小鼠黑色素瘤B16F10细胞增殖机制研究[J].中国药理学通报,2015,31(7):967-72.
[8]Wang Y M,Liu Y,Yan X Y,et al.Inhibitory effects of Licochalcone A on proliferation of melanoma B16F10 cells[J].Chin Pharmacol Bull,2015,31(7):967-72.
[9]Chu X,Ci X,Wei M,et al.Licochalcone A inhibits lipopolysaccharide-induced inflammatory response in vitro and in vivo[J].J Agric Food Chem,2012,60(15):3947-54.
[10]Furusawa J,Funakoshi-Tago M,Tago K,et al.Licochalcone A significantly suppresses LPS signaling pathway through the inhibition of NF-kappaB p65 phosphorylation at serine 276[J].Cell Signal,2009,21(5):778-85.
[11]Shen L L,Liu Y N,Shen H J,et al.Inhalation of glycopyrronium inhibits cigarette smoke-induced acute lung inflammation in a murine model of COPD[J].Int Immunopharmacol,2014,18(2):358-64.
[12]Li F F,Shen J,Shen H J,et al.Shp2 plays an important role in acute cigarette smoke-mediated lung inflammation[J].J Immunol,2012,189(6):3159-67.
[13]Liao S X,Ding T,Rao X M,et al.Cigarette smoke affects dendritic cell maturation in the small airways of patients with chronic obstructive pulmonary disease[J].Mol Med Rep,2015,11(1):219-25.
[14]Thatcher T H,McHugh N A,Egan R W,et al.Role of CXCR2 in cigarette smoke-induced lung inflammation[J].Am J Physiol Lung Cell Mol Physiol,2005,289(2):L322-8.
[15]Pelegrino N R,Tanni S E,Amaral R A,et al.Effects of active smoking on airway and systemic inflammation profiles in patients with chronic obstructive pulmonary disease[J].Am J Med Sci,2013,345(6):440-5.
[16]Eltom S,Belvisi M G,Stevenson C S,et al.Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation:an insight into the pathogenesis of COPD[J]. PLoS One,2014,9(11):e112829.
[17]Culpitt S V,Rogers D F,Shah P,et al.Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease[J].Am J Respir Crit Care Med,2003,167(1):24-31.
[18]Taylor M,Vella L,Carr T.Assessment of an in vitro model of lung epithelial cell stress responses[J].Free Radic Biol Med,2014,75(1):S51.
[19]郭峰,况九龙.超氧化物歧化酶基因多态性及其活力与慢性阻塞性肺疾病的易感性研究[J].中华结核和呼吸杂志,2011,34(6):424-8.
[19]Guo F,Kuang J L.Superoxide dismutase gene polymorphisms and functional activity in chronic obstructive pulmonary disease[J]. Chin J Tuberc Respir Dis,2011,34(6):424-8.
[20]Lv H,Ren H,Wang L,et al.Lico A enhances Nrf2-mediated defense mechanisms against t-BHP-induced oxidative stress and cell death via Akt and ERK activation in RAW 264.7 cells[J].Oxid Med Cell Longev,2015,2015:709845.
[21]Fernandes D J,Bonacci J V,Stewart A G.Extracellular matrix,integrins,and mesenchymal cell function in the airways[J].Curr Drug Targets,2006,7(5):567-77.
[22]Gueders M M,Foidart J M,Noel A,et al.Matrixmetalloproteinases(MMPs)and tissue inhibitors of MMPs in the respiratory tract:potential implications in asthma and other lung diseases[J].Eur J Pharmacol,2006,533(1-3):133-44.
[23]Nakamura M,Wada H,Honda K,et al.Clarithromycin ameliorates pulmonary inflammation induced by short term cigarette smoke exposure in mice[J].Pulm Pharmacol Ther,2015,35:60 -6.
[24]Srivastava P K,Dastidar S G,Ray A.Chronic obstructive pulmonary disease:role of matrix metalloproteases and future challenges of drug therapy[J].Expert Opin Investig Drugs,2007,16(7):1069-78.
[25]Lee K H,Lee C H,Jeong J,et al.Neutrophil elastase differentially regulates interleukin 8(IL-8)and vascular endothelial growth gactor(VEGF)production by cigarette smoke extract[J].J Biol Chem,2015,290(47):28438-45.
[26]Larsson K.Inflammatory markers in COPD[J].Clin Respir J,2008,2(1):84-7.
[27]Schneider D,Ganesan S,Comstock A T,et al.Increased cytokineresponse of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease[J].Am J Respir Crit Care Med,2010,182(3):332-40.
[28]Shin Y O,Park C H,Lee G H,et al.Heat-processed scutellariae radix enhances anti-inflammatory effect against lipopolysaccharideinduced acute lung injury in mice via NF-κB signaling[J].Evid Based Complement Alternat Med,2015,2015:456846.
[29]刘丹丹,曹纲,张琦,等.三叶青黄酮经p38MAPK和NF-κB途径抑制老年小鼠急性肺损伤[J].中国药理学通报,2015,31(12):1725-9.
[29]Liu D D,Cao G,Zhang Q,et al.Inhibition effect of flavonoids from Radix tetrastigmae on acute lung injury of aged mice through p38MAPK and NF-κB pathawy[J].Chin Pharmacol Bull,2015, 31(12):1725-9.
[30]Liu Y,Liu Y,Xu D,Li J.Latanoprost-induced cytokine and chemokine release from human tenon's capsule fibroblasts:role of MAPK and NF-κB signaling pathways[J].J Glaucoma,2015,24(9):635-41.
[31]Bao M J,Shen J,Jia Y L,et al.Apple polyphenol protects against cigarette smoke-induced acute lung injury[J].Nutrition,2013,29(1):235-43.
[32]Lin X X,Yang X F,Jiang J X,et al.Cigarette smoke extract-induced BEAS-2B cell apoptosis and anti-oxidative Nrf-2 up-regulation are mediated by ROS-stimulated p38 activation[J].Toxicol Mech Methods,2014,24(8):575-83.
Licochalcone A protects against cigarette smoke-mediated acute lung injury in mice by suppressing ERK1/2/NF-κB pathways
REN Qian-qian1,WANG Li-pei2,ZHAO Wei3,LU Hong1,XIE Qiang-min4,ZHANG Shui-juan1
(1.College of Pharmaceutical Science,2.College of Basic Medical Science,Zhejiang Chinese Medical University,Hangzhou310053,China;
3.the First Affiliated Hospital of Zhejiang Chinese Medical University,Hangzhou310006,China;4.Zhejiang Respiratory Drugs Research Laboratory,Zhejiang University School of Medicine,Hangzhou310058,China)
AimTo explore the protective roles of licochalcone A(LA)on mice with cigarette smoke-mediated acute lung injury and the related mechanisms. MethodsIn vivo:Mice were exposed to cigarette smoke(CS)to establish acute lung injury model.The bronchoalveolar lavage fluid(BALF)was conducted for cell counting.The mRNA and protein expression of keratinocyte-derived chemokine(KC),tumor necrosis factor alpha(TNF-α),interleukin 1β(IL-1β)and matrix metalloproteinases(MMP)-9 in lungs were determined.The myeloperoxidase(MPO),superoxide dismutase(SOD)activities and glutathione(GSH)levels in lungs were quantified.The paraffin sections of lungs were prepared and stained with HE.In vitro:Human lung epithelial cells(BEAS-2B)were exposed to cigarette smoke extract(CSE),which induced cell injury.The releases of interleukin 8(IL-8)and MMP-9 were assessed.The phosphorylation of mitogen-activated protein kinases(MAPKs,including ERK1/2,p38 and JNK)and nuclear factor-κB(NF-κB)p65 protein were analyzed by Western blot.ResultsIn vivo:The accumulation of inflammatory cells was lower in LA groups than that in model group.In comparison with normal control group,the mRNA and protein lev-els of KC,TNF-α,IL-1β and MMP-9 were significantly increased in model group.Following treatment with LA,the above indicators were significantly decreased as compared to model group.In the CS-exposed mice,the MPO activity in lungs was significantly increased,meanwhile the SOD activity and GSH level were significantly decreased compared with the air-exposed animals.CS-induced activity of MPO was significantly inhibited,which were accompanied by increases in SOD and GSH levels by LA.In vitro:CSE-induced mRNA levels of IL-8 and MMP-9 were significantly inhibited by LA at 2.5 and 5 μmol·L-1.The CSE-induced phosphorylation of ERK1/2 and nucleus NF-κB p65 protein expression were prevented by pretreatment with LA.ConclusionsLA has protective effects on CS-exposed acute lung injury in mice by preventing CS-induced pulmonary inflammation,oxidative stress and protease rise.The exploration of the mechanisms suggests that LA exerts protective effects via suppressing ERK1/2/NF-κB pathways.
licorice;licochalcone A;lung injury;cigarette smoke;lung epithelial cell;cytokines;signal pathway
2016-01-07,修回日期:2016-02-02
浙江省教育厅科研项目(No Y201328230);浙江中医药大学校级科研基金项目(No 2010ZY07)
任倩倩(1989-),女,硕士生,研究方向:抗炎免疫药理学,E-mail:61324680@qq.com;张水娟(1980-),女,博士,实验师,研究方向:抗炎免疫药理学,通讯作者,E-mail:zsjdxw@163.com
10.3969/j.issn.1001-1978.2016.05.011
A
1001-1978(2016)05-0643-09
R-332;R284.1;R163.1;R322.35;R563.902.5
