第一性原理研究混合钙钛矿CH3NH3PbxSn1-xI3结构及光电特性
2016-09-09鲁效庆赵兹罡李可魏淑贤瞿媛媛牛永强刘学锋
鲁效庆 赵兹罡 李可 魏淑贤 瞿媛媛 牛永强 刘学锋
(中国石油大学(华东)理学院,山东青岛266580)
第一性原理研究混合钙钛矿CH3NH3PbxSn1-xI3结构及光电特性
鲁效庆*赵兹罡李可魏淑贤瞿媛媛牛永强刘学锋*
(中国石油大学(华东)理学院,山东青岛266580)
有机/无机钙钛矿是一类极具潜质的光电材料,目前已实现超过20%的光电转化效率。本文采用第一性原理对有机/无机混合钙钛矿CH3NH3PbxSn1-xI3(x=0-1)的结构及光电特性进行了理论研究。结果表明,范德华力(VDW)在优化钙钛矿结构中起着重要的作用,考虑范德华力可减小Pb/Sn―I键长,从而减小体系体积。通过分析甲胺离子CH3NH+3的态密度和Bader电荷,我们发现其对前线轨道没有贡献,仅仅扮演电荷供体的角色。Pb/Sn与I之间同时存在共价键和离子键相互作用。价带顶(VBM)主要是由I 5p以及Pb 6s(Sn 5s)杂化组成,而导带底(CBM)主要由Pb 6p(Sn 5p)轨道组成。在可见光区,随着波长的增加,体系吸收强度呈现整体下降趋势;随着Sn/Pb比值逐渐增大,吸收强度呈现增大趋势。CH3NH3SnI3在可见光区表现出较佳的吸收光谱特性。
钙钛矿;光电特性;能带结构;态密度;介电函数;第一性原理
1 引言
有机/无机混合钙钛矿(ABX3)太阳能电池,因其生产工艺简单,成本低廉,光电转化效率较高而引起了广泛关注1。2009年,Miyasaka等2首次将钙钛矿CH3NH3PbX3(X=I,Br)应用到染料敏化太阳能电池,取得了3.8%的光电转化效率。此后,钙钛矿太阳能电池获得飞速发展,截止目前钙钛矿太阳能电池的最高认证效率已突破20.0%3-7。
CH3NH3PbI3钙钛矿研究最为广泛,但最近研究表明,混合钙钛矿(包括A、B、X混合)展现出优异的光电特性。对于A混合,Grätzel等8通过甲脒(CHNH3+,FA)和甲胺(CH3NH3+,MA)混合得到FAxMA1-xPbI3(x=0-1)钙钛矿,相比于MAPbI3,此混合钙钛矿能够显著提高电池的短路电流。Snaith等9发现在混合钙钛矿CH3NH3PbI3-xClx(x= 0-3)中,电子-空穴对的输运长度超过了1µm。然而,对于B混合钙钛矿,研究相对较少。实验研究证实,Sn基钙钛矿具有较广的光谱响应范围,能够拓展至红外光区10,11。Kanatzidis等12制作了CH3NH3SnI3钙钛矿太阳能电池,并且采用spiro-OMeTAD作为空穴传输层,拓展钙钛矿太阳能电池吸光范围至950 nm,相比CH3NH3PbI3太阳能电池发生了显著红移。同时他们合成了一系列混合CH3NH3Sn1-xPbxI3钙钛矿13,证明了其在700-1000 nm范围内具有较强的光电响应。Hayase等14报道了全固态Sn/Pb基混合钙钛矿太阳能电池,发现CH3NH3Sn0.5Pb0.5I3具有最佳的光电特性,吸收光谱拓展至1060 nm。然而,对于混合钙钛矿CH3NH3PbxSn1-xI3(x=0-3)的结构特点、电子结构及内部光电转化过程的理论研究相对较少。为深入认识混合钙钛矿的光电转化机理,我们采用第一性原理研究了混合钙钛矿CH3NH3PbxSn1-xI3(x= 0-1)的微观结构、电子及光谱特性。
2 计算方法
本文所有量化计算基于第一性原理的VASP (Vienna ab-initio simulation package)软件包,采用投影缀加平面波(PAW)赝势完成,其中在处理电子间相互作用的交换关联能时采用广义梯度近似(GGA)PBE泛函15,16。体系结构优化时晶格常数和所有原子设置为全面弛豫,对体系对称性未加以限制。在计算中平面波截断能选取为Ecut=500 eV,布里渊区积分采用4×4×4的Monkhorst-Pack K点网格17,高斯展宽因子为0.05,能量收敛标准为10-5eV∙atom-1。为验证范德华力在优化体系构型中起的作用,本文采用PAW-PBE计算方法,并在此方法基础上考虑范德华力(VDW)校正,分别进行结构优化。在CH3NH3PbI3体系中,由于Pb具有较强的相对论效应,存在自旋轨道耦合效应(SOC)18。然而前期研究表明,采用GGA和无SOC计算方法得到的禁带宽度也相对比较准确19。因此,所选择方法基组可较准确地描述CH3NH3PbxSn1-xI3(x=0-1)钙钛矿的结构及禁带宽度等。
3 结果与讨论
3.1结构特性有机/无机混合钙钛矿通常由阳离子A(A= CH3NH3+,HC(NH2)2+)和八面体BX6(B=Si2+,Ge2+,Pb2+,Sn2+;X=Cl-,Br-,I-)组成。图1为混合钙钛矿CH3NH3Pb0.5Sn0.5I3结构示意图,其中Pb(Sn)与周围6个I离子成键形成八面体构型,CH3NH+3位于八面体间隙中心,半径RA=0.18 nm20,配位数为12。在CH3NH3SnxPb1-xI3中,平面内相邻甲胺离子的C―N键呈垂直排布,Pb―I键长范围为0.314-0.322 nm,而Sn―I键长范围为0.307-0.318 nm。Goldschmidt21曾提出容忍因子t来描述钙钛矿结构稳定性与离子半径之间的关系:

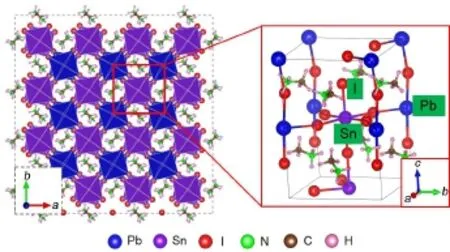
图1 混合钙钛矿CH3NH3Pb0.5Sn0.5I3结构示意图Fig.1 Schematic structure of mixed perovskite CH3NH3Pb0.5Sn0.5I3
其中RA、RB和RX分别是A、B和X离子的半径。一般而言,当t接近1时,钙钛矿构型呈现理想的立方结构(空间群Pm3m)。当t偏离1较大时,构型将会发生晶体形变。考虑到Pb2+、I-的离子半径分别为0.12、0.22 nm,CH3NH3PbI3的t值为0.83。由于Sn2+的离子半径(0.11 nm)小于Pb2+,因此用Sn代替Pb可以提高t值,进而增强体系的稳定性。常温下CH3NH3PbI3呈四方晶型(空间群I4/m),因此本文研究对象设定为四方型混合钙钛矿CH3NH3PbxSn1-xI3(x=0-1)。其中,我们构建x值为0、0.25、0.5、0.75和1五种结构模型进行结构优化和性质分析。
在PAW-PBE方法基础上,考虑VDW校正,对初始构型进行优化,发现范德华力对钙钛矿构型有较大影响。优化后的混合CH3NH3PbxSn1-xI3的晶格常数如表1所示,计算结果与实验和计算值(见表1注释)13,22,23吻合较好。范德华力通过作用于体系中的I―H、Pb/Sn―I键,使得Pb/Sn―I键长缩短明显,减小了体系的体积并增强了其稳定性,例如,CH3NH3PbI3在VDW校正后Pb―I键长由0.325 nm缩短至0.318 nm;CH3NH3SnI3在VDW校正后Sn―I键长由0.315 nm缩短至0.313 nm。随着Pb含量的增加,CH3NH3PbxSn1-xI3晶胞的体积呈增大趋势。
3.2电子特性
基于稳定构型基础上,考虑到混合钙钛矿电子特性的相似性,我们选取CH3NH3PbI3为例绘制总态密度(TDOS)和分波态密度(PDOS)图,见图2。由总态密度图可以得出CH3NH3PbI3禁带宽度Eg为1.55 eV,与实验值1.55 eV6、1.58 eV24基本吻合,这也验证了我们所选方法的准确性。通过分析CH3NH+3态密度,发现CH3NH+3在费米能级附近没有明显的态密度分布,表明CH3NH+3发生受光电子跃迁几率较小。CH3NH3+与I-没有明显耦合作用,证明CH3NH+3与I-之间为离子键相互作用。通过分析Pb和I的分波态密度,发现价带顶(VBM)主要由Pb 6s(Sn 5s)和I 5p轨道杂化的反键轨道组成,而导带底(CBM)主要由Pb 6p(Sn 5p)轨道组成。表2列出了CH3NH3PbxSn1-xI3沿Γ(0,0,0)→Z(0,0,0.5)路径的电子空穴有效质量。从表中数据可知:载流子有效质量较小,利于电荷传输,并且随着Sn/ Pb比值的增大,电子空穴有效质量逐渐减小。这与他人研究结果一致,即s-p杂化可以产生相对较小的电子空穴有效质量25,从而产生较大的电子输运长度。
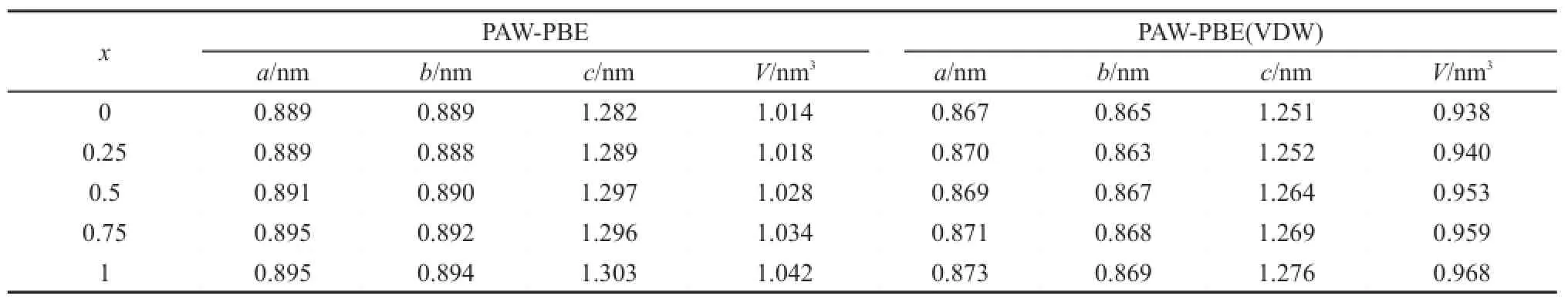
表1 PAW-PBE和PAW-PBE(VDW)优化后CH3NH3PbxSn1-xI3晶格常数和体积Table 1Lattice constants and volumes of CH3NH3PbxSn1-xI3calculated by PAW-PBE without and with VDW correction
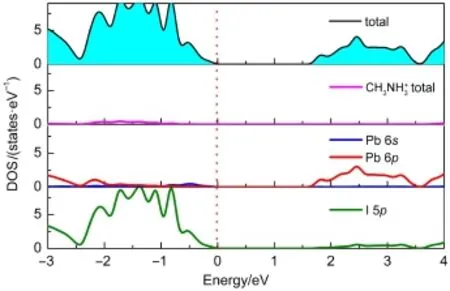
图2 CH3NH3PbI3的总态密度和分波态密度Fig.2Total density of state(DOS)and partial density of state of CH3NH3PbI3
图3描绘了CH3NH3Pb0.5Sn0.5I3在Γ点价带顶和导带底的可视化波函数。在价带顶处,可视化波函数(即电子出现的几率)主要聚集在Pb/Sn和I周围,而在导带底处,可视化波函数主要围绕在Pb/ Sn周围。这进一步验证了电子被激发时将主要由Pb 6s(Sn 5s)和I 5p轨道跃迁至Pb 6p(Sn 5p)轨道。
钙钛矿禁带宽度对于吸收光谱范围具有决定性意义。图4(a)所示为CH3NH3PbxSn1-xI3体系能带结构图,范围为-1.5-2.5 eV。由图可知,混合钙钛矿具有相似的能带结构,它们的价带顶和导带底都位于布里渊区的同一Γ(0,0,0)对称点,表明此钙钛矿体系是直接带隙半导体,利于电子吸收光子发生垂直跃迁,降低不必要的能量损失,提高光电转换效率。导带底波动相对平缓,表明电子轨道能级是非局域的,因此电子在钙钛矿内部可以进行长距离输运。图4(b)为CH3NH3PbxSn1-xI3态密度图,当混合钙钛矿中Sn/Pb的比例逐渐增大时,禁带宽度随之减小。因为价带顶主要是由Pb 6s(Sn 5s)-I 5p轨道杂化的反键态组成,Sn 5s轨道能级高于Pb 6s轨道能级,因此Sn 5s-I 5p轨道之间的耦合强度高于Pb 6s-I 5p,进而抬高了VBM的能级,减小了禁带宽度。为了便于直观地查看和比较不同Pb/Sn混合比例钙钛矿的禁带宽度值,并将能带结构图与态密度图对应,我们将图4能带结构和态密度图中费米能级设定为各钙钛矿价带顶值,因此VBM的升高表现为CBM的降低。图4(c)显示为CH3NH3PbxSn1-xI3空间电荷密度分布图,可以发现Pb/Sn―I之间有明显的电子耦合,表明他们之间存在共价键相互作用,而CH3NH3+与I-之间没有明显电子耦合,表明它们之间存在离子键相互作用。
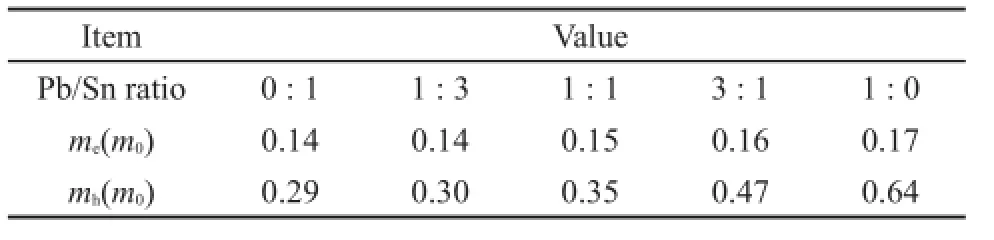
表2 CH3NH3PbxSn1-xI3沿Γ(0,0,0)→Z(0,0,0.5)路径的载流子有效质量Table 2Carriers′effective mass of CH3NH3PbxSn1-xI3along Γ(0,0,0)→Z(0,0,0.5)path
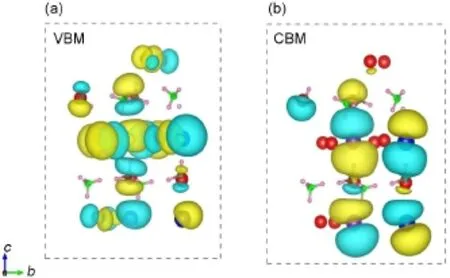
图3 MAPb0.5Sn0.5I3在Γ位置的可视化波函数Fig.3Visualization wave functions of MAPb0.5Sn0.5I3at the Г point(a)VBM(valence band maximum),(b)CBM(conduction band minimum)
表3列出了钙钛矿体系Bader电荷值。表3数据显示Pb/Sn和I的Bader电荷与通常的纯离子键相互作用(例如Pb2+、Sn2+、I-)偏离较大,证明了Pb/ Sn2+与I-之间不仅存在共价键作用而且还有离子键相互作用。Pb/Sn―I共价相互作用也可以由图2得到证实,Pb 6s,6p(Sn 5s,5p)轨道与I 5p轨道有明显的耦合作用。CH3NH3+离子对于前线轨道没有显著贡献,它们只作为电荷供体为Pb/SnI3骨架提供大约0.7e电荷。与C相连的H和与N相连的H是不对等的,CH3相对NH3来说,为Pb/SnI3骨架提供了更多的电荷。
3.3光谱特性
介电函数是反映钙钛矿光谱特性的有效指标之一,用以描述半导体电磁辐射的线性响应属性。介电函数的虚部ε2(ω)源于选择定则下的占据和未占据波函数间的动量矩阵元,而实部ε1(ω)可以借助Kramer-Kronig关系式26由虚部ε2(ω)获得。因此,虚部ε2(ω)对于洞察电子跃迁规律和吸光特性有重要意义。图5(a)描述了CH3NH3PbxSn1-xI3(x= 0,0.25,0.5,0.75,1)体系的介电特性,图中不同的峰值对应于特定的轨道电子跃迁。在380 nm附近均出现了波峰,此波峰主要对应于Pb 6s和I 5p之间的杂化轨道到Pb 6p轨道的电子跃迁,且峰值随Sn/Pb比值增大而降低,此过程与图4(b)态密度吻合,出现在价带顶的波峰和导带底的波峰之间的能量差均为3.3 eV左右,此能量对应于吸收光波长380 nm。在长波长区,CH3NH3PbxSn1-xI3波峰随着Sn/Pb比值的减小而逐渐降低并发生蓝移。在680 nm处,CH3NH3SnI3出现了一个最高峰值,此波峰对应的电子跃迁能级差约为1.8 eV;在510 nm处出现另外一个较大的波峰,表明CH3NH3SnI3在该波长范围具有更佳的吸收特性。
钙钛矿吸收系数I(ω)可通过介电函数的实部ε1(ω)和虚部ε2(ω)求得27:
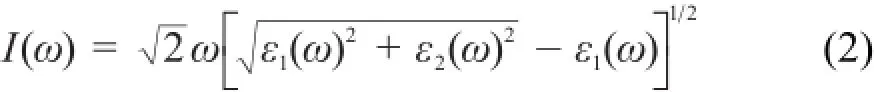
由图5(b)显示的混合钙钛矿体系的吸收系数I(ω)曲线可以看出:在350 nm处CH3NH3PbxSn1-xI3吸收强度最大,其中CH3NH3PbI3的峰值相对较高,CH3NH3PbxSn1-xI3峰值随着Sn/Pb比值增大略有降低。在400-800 nm,随着波长的增加,体系吸收强度呈现整体下降的趋势;随着Sn/Pb比值逐渐增大吸收强度呈增大趋势。整体而言,相对于其他混合CH3NH3PbxSn1-xI3钙钛矿,CH3NH3SnI3在可见光区具有较佳的光谱吸收特性。但对于CH3NH3SnI3,存在Sn容易氧化的困扰13,28,因此如何控制减缓其氧化速率也是今后研究的重点之一。
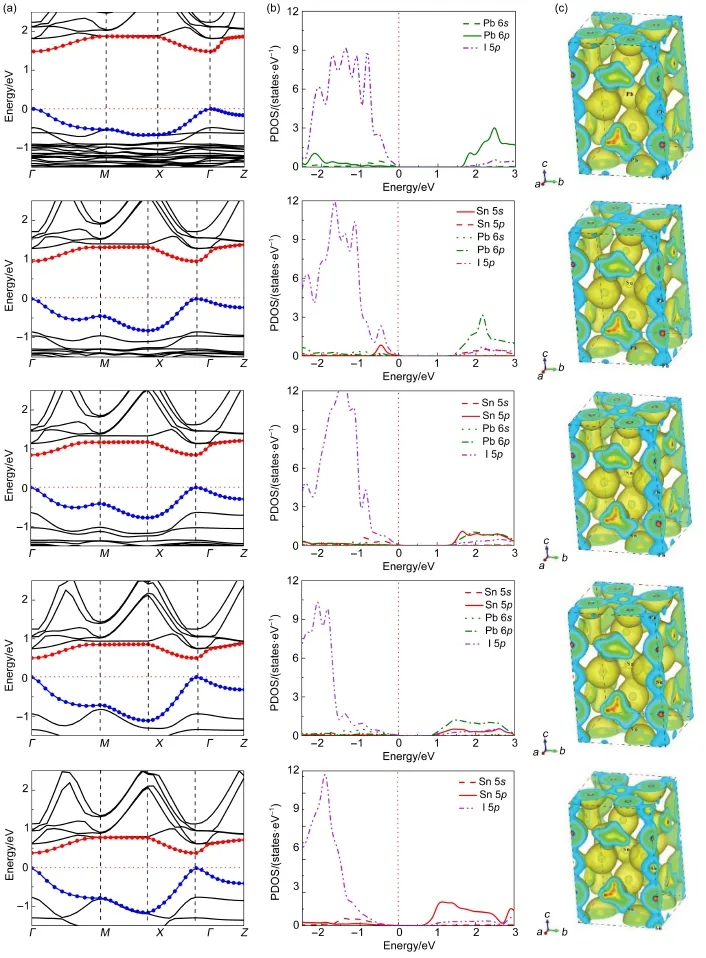
图4 CH3NH3PbxSn1-xI3(x=0,0.25,0.5,0.75,1;自下而上)的(a)能带结构图,(b)态密度和(c)电荷密度分布图Fig.4(a)Band structure,(b)density of state,and(c)charge density of CH3NH3PbxSn1-xI3(x=0,0.25,0.5,0.75,1,from bottom to top)

表3 CH3NH3Pb0.5Sn0.5I3的Bader电荷值Table 3Bader charges of CH3NH3Pb0.5Sn0.5I3
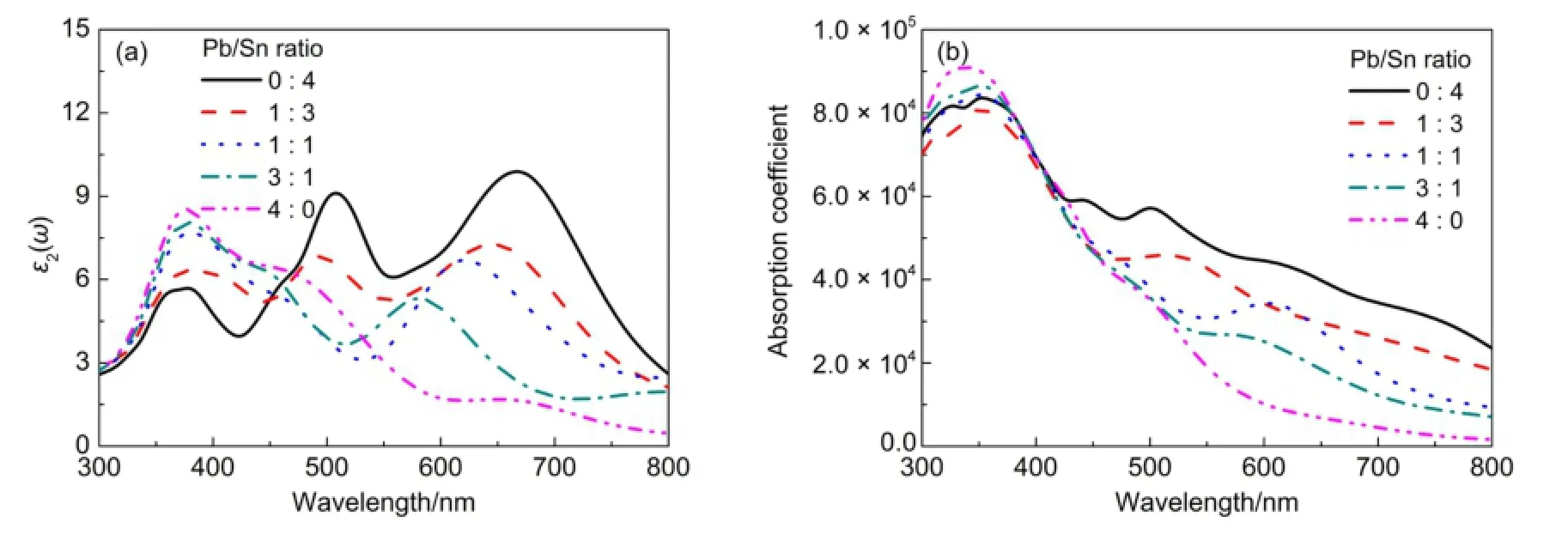
图5 (a)混合钙钛矿介电函数(虚部ε2(ω))及(b)吸收光谱图Fig.5(a)Imaginary part of dielectric function(ε2(ω))and(b)absorption spectra of mixed perovskite
4 结论
本文运用第一性原理对CH3NH3PbxSn1-xI3(x= 0,0.25,0.5,0.75,1)混合钙钛矿体系的微观结构和光电特性进行理论研究。结果发现,CH3NH+3在调控钙钛矿结构中起重要的作用,其本身不直接参与前线轨道的电子跃迁过程,仅作为电荷供体为Pb/SnI3骨架提供大约0.7e电荷,CH3相对NH3来说,为Pb/SnI3骨架提供了更多的电荷。CH3NH+3与I-之间是离子键相互作用,而Pb/Sn2+与I-之间同时存在共价键和离子键相互作用。受光激发后,钙钛矿内部的电子将由价带顶的I 5p和Pb 6s(Sn 5s)轨道杂化的反键态跃迁至导带底的Pb 6p(Sn 5p)轨道。随着Sn/Pb比值的增大,混合钙钛矿的晶格常数和体积逐渐减小;电子空穴有效质量呈现逐渐减小趋势;禁带宽度值逐渐变小;吸收光谱发生红移,在可见光区强度呈增大趋势。结果表明CH3NH3SnI3更具有作为钙钛矿电池吸光层的潜质。
References
(1)Service,R.F.Science 2014,344(6183),458.doi:10.1126/ science.344.6183.458
(2)Kojima,A.;Teshima,K.;Shirai,Y.;Miyasaka,T.J.Am.Chem. Soc.2009,131(17),6050.doi:10.1021/ja809598r
(3)Im,J.H.;Lee,C.R.;Lee,J.W.;Park,S.W.;Park,N.G. Nanoscale 2011,3(10),4088.doi:10.1039/C1NR10867K
(4)Lee,M.M.;Teuscher,J.;Miyasaka,T.;Murakami,T.N.;Snaith,H.J.Science 2012,338(6107),643.doi:10.1126/ science.1228604
(5)Burschka,J.;Pellet,N.;Moon,S.J.;Humphry-Baker,R.;Gao,P.;Nazeeruddin,M.K.;Grätzel,M.Nature 2013,499(7458),316.doi:10.1038/nature12340
(6)Zhou,H.P.;Chen,Q.;Li,G.;Luo,S.;Song,T.B.;Duan,H.S.;Hong,Z.R.;You,J.B.;Liu,Y.S.;Yang,Y.Science 2014,345 (6196),542.doi:10.1126/science.1254050
(7)Jeon,N.J.;Noh,J.H.;Yang,W.S.;Kim,Y.C.;Ryu,S.;Seo,J.;Seok,S.I.Nature 2015,517(7535),476.doi:10.1038/ nature14133
(8)Pellet,N.;Gao,P.;Gregori,G.;Yang,T.Y.;Nazeeruddin,M. K.;Maier,J.;Grätzel,M.Angew.Chem.Int.Edit.2014,53(12),3151.doi:10.1002/anie.201309361
(9)Stranks,S.D.;Eperon,G.E.;Grancini,G.;Menelaou,C.;Alcocer,M.J.;Leijtens,T.;Herz,L.M.;Petrozza,A.;Snaith,H. J.Science 2013,342(6156),341.doi:10.1126/science.1243982
(10)Noel,N.K.;Stranks,S.D.;Abate,A.;Wehrenfennig,C.;Guarnera,S.;Haghighirad,A.A.;Sadhanala,A.;Eperon,G.E.;Pathak,S.K.;Johnston,M.B.;Petrozza,A.M.;Herz,L.M.;Snaith,H.J.Energy Environ.Sci.2014,7(9),3061. doi:10.1039/C4EE01076K
(11)Chung,I.;Song,J.H.;Im,J.;Androulakis,J.;Malliakas,C.D.;Li,H.;Freeman,A.J.;Kenney,J.T.;Kanatzidis,M.G. J.Am.Chem.Soc.2012,134(20),8579.doi:10.1021/ja301539s
(12)Hao,F.;Stoumpos,C.C.;Cao,D.H.;Chang,R.P.;Kanatzidis,M.G.Nat.Photon.2014,8(6),489.doi:10.1038/ nphoton.2014.82
(13)Stoumpos,C.C.;Malliakas,C.D.;Kanatzidis,M.G.Inorg. Chem.2013,52(15),9019.doi:10.1021/ic401215x
(14)Ogomi,Y.;Morita,A.;Tsukamoto,S.;Saitho,T.;Fujikawa,N.;Shen,Q.;Toyoda,T.;Yoshino,K.;Pandey,S.S.;Ma,T.;Hayase,S.J.Phys.Chem.Lett.2014,5(6),1004. doi:10.1021/jz5002117
(15)Kresse,G.;Furthmüller,J.Comp.Mater.Sci.1996,6(1),15. doi:10.1016/0927-0256(96)00008-0
(16)Kresse,G.;Joubert,D.Phys.Rev.B 1999,59(3),1758. doi:10.1103/PhysRevB.59.1758
(17)Monkhorst,H.J.;Pack,J.D.Phys.Rev.B 1976,13(12),5188. doi:10.1103/PhysRevB.13.5188
(18)Even,J.;Pedesseau,L.;Jancu,J.M.;Katan,C.J.Phys.Chem. Lett.2013,4(17),2999.doi:10.1021/jz401532q
(19)Mosconi,E.;Amat,A.;Nazeeruddin,M.K.;Grätzel,M.;De Angelis,F.J.Phys.Chem.C 2013,117(27),13902. doi:10.1021/jp4048659
(20)McKinnon,N.K.;Reeves,D.C.;Akabas,M.H.J.Gen.Phys. 2011,138(4),453.doi:10.1085/jgp.201110686
(21)Goldschmidt,V.M.Sci.Nat.1926,14(21),477.doi:10.1007/ BF01507527
(22)Zhu,X.;Su,H.;Marcus,R.A.;Michel-Beyerle,M.E.J.Phys. Chem.Lett.2014,5(17),3061.doi:10.1021/jz501174e
(23)Baikie,T.;Fang,Y.;Kadro,J.M.;Schreyer,M.;Wei,F.;Mhaisalkar,S.G.;Grätzel,M.;White,T.J.J.Mater.Chem.A 2013,1(18),5628.doi:10.1039/C3TA10518K
(24)Ding,X.K.;Li,X.M.;Gao,X.D.;Zhang,S.D.;Huang,Y.D.;Li,H.R.Acta Phys.-Chim.Sin.2015,31(3),576.[丁绪坤,李效民,高相东,张树德,黄宇迪,李浩然.物理化学学报,2015,31(3),576.]doi:10.3866/PKU.WHXB201501201
(25)Yin,W.J.;Shi,T.T.;Yan,Y.F.Appl.Phys.Lett.2014,104(6),063903.doi:10.1063/1.4864778
(26)Sun,J.;Wang,H.T.;He,J.L.;Tian,Y.J.Phys.Rev.B 2005,71 (12),125132.doi:10.1103/PhysRevB.71.125132
(27)Shi,H.L.;Chu,M.F.;Zhang,P.J.Nucl.Mater.2010,400(2),151.doi:10.1016/j.jnucmat.2010.02.024
(28)Grätzel,M.Nat.Mater.2014,13(9),838.doi:10.1038/ nmat4065
First-Principles Investigation of the Structural and Photoelectronic Properties of CH3NH3PbxSn1-xI3Mixed Perovskites
LU Xiao-Qing*ZHAO Zi-GangLI KeWEI Shu-Xian QU Yuan-YuanNIU Yong-QiangLIU Xue-Feng*
(College of Science,China University of Petroleum,Qingdao 266580,Shandong Province,P.R.China)
Organic/inorganic perovskites have exhibited great potential as photoelectronic materials,achieving remarkable photoelectric conversion efficiency,currently over 20%.The structural,electronic,and optical properties of organic/inorganic hybrid CH3NH3PbxSn1-xI3perovskites(x=0-1)have been investigated by the first-principles theory.Our results indicate that the van der Waals(VDW)interaction plays a crucial role in the structure optimization.Accounting for VDW force correction,both the Pb/Sn―I bond lengths and volumes are decreased.By analyzing the density of states and the Bader charge of CH3NH3+cations,we find that cations contribute only slightly to the band edge,but play the role of charge donors.There exists a combined covalent and ionic interaction between Pb/Sn and I ions.The valence band maximum(VBM)is mainly contributed by the I 5p orbitals with the overlapping of Pb 6s(Sn 5s)orbitals,while the conduction band minimum(CBM)is dominated by Pb 6p(Sn 5p)orbitals.In the visible light region,with increasing wavelength,the absorption intensity demonstrates a decreasing trend;as the Sn/Pb ratio increases,the absorption intensity shows an increasing trend.CH3NH3SnI3perovskites demonstrate great potential to absorb light in the visible region.
Perovskite;Photoelectric property;Band structure;Density of states;Dielectric function;First-principles theory
January 6,2016;Revised:March 14,2016;Published on Web:March 15,2016.
O641
[Article]10.3866/PKU.WHXB201603154www.whxb.pku.edu.cn
*Corresponding authors.LU Xiao-Qing,Email:luxq@upc.edu.cn;Tel:+86-532-86983372.LIU Xue-Feng,Email:liuxf@upc.edu.cn.
The project was supported by the National Natural Science Foundation of China(21303266)and Fundamental Research Funds for the Central Universities,China(15CX05050A,14CX02214A).
国家自然科学基金(21303266)和中央高校自主创新项目(15CX05050A,14CX02214A)资助
©Editorial office ofActa Physico-Chimica Sinica
