贵金属原子掺杂的CeO2(111)表面模型催化剂的理论研究
2016-09-02崔鹏飞金丽芳张建旺袁金焕赵雷洪
崔鹏飞,金丽芳,张建旺,袁金焕,赵雷洪
(浙江师范大学物理化学研究所,先进催化材料教育部重点实验室,浙江 金华 321004)
贵金属原子掺杂的CeO2(111)表面模型催化剂的理论研究
崔鹏飞,金丽芳,张建旺,袁金焕,赵雷洪
(浙江师范大学物理化学研究所,先进催化材料教育部重点实验室,浙江金华321004)
通过DFT(密度泛函理论)的方法,详细的计算了不同贵金属原子,金(Au)、钯(Pd)、铂(Pt)与铑(Rh)在CeO2(111)表面掺杂模型催化剂的几何结构与电子性质情况。计算表明:掺杂贵金属原子将会使CeO2(111)几何构型发生不同程度的改变,将贵金属金、钯、铂与铑掺杂到二氧化铈中,形成模型催化剂。该类催化剂的氧空位形成能为0.32、0.41、1.04和1.42 eV比体相CeO2(111)表面的氧空位形成能大幅减小,有利于促进了氧空位的形成,掺杂体系的催化活性得到大幅提高。对态密度计算结果进行分析,在二氧化铈体相中掺杂贵金属原子,在费米能级处出现所掺杂的贵金属的电子锋,这充分说明,贵金属掺杂与CeO2(111)的相互作用明显大于贵金属吸附与CeO2(111)的相互作用。依照Au、Pd、Pt和Rh的次序,在CeO2(111)面掺杂金属原子失去电子的数目在明显的增多,在CeO2(111)面出现氧空位时,掺杂金属原子的电荷比相应化学计量模型电荷低。
密度泛函理论;二氧化铈;贵金属;电子态密度;掺杂
金属负载在氧化铈载体上的铈基催化剂在汽车尾气处理、挥发性有机物催化消除、燃料电池等方面具有广泛的应用[1-3]。众多研究结果表明,金属与CeO2载体间的相互作用是影响催化剂活性与稳定性的关键因素。一般来说,贵金属与二氧化铈载体之间的作用方式主要分为两种:第一种金属以纳米粒子的形式均匀的分散在载体表面,形成最为常见的负载类型催化剂(Mx/CeO2);第二种是金属原子进入二氧化铈的晶格内部,取代铈原子,形成取代型金属铈基催化剂(MxCe1-xO2-δ)。从催化剂的制备方法来看,制备的催化体系中两种相互作用方式很可能并存。袁金焕等[4]着重考察了了不同贵金属原子在CeO2(111)表面的吸附情况,结果表明当金属原子的吸附在CeO2(111)表面,可以有效的促进Ce4+的还原,从而形成低价态的Ce3+离子,对后续形成活性超氧物种具有重要作用。
另一方面,大量的实验结果表明,将贵金属掺杂在二氧化铈表面,从而形成取代型复合氧化物,同样可以在一定程度上提高CeO2的催化活性。目前,系统比较不同贵金属掺杂在CeO2催化剂的结构及电子性质的理论研究较少[5]。本文采用DFT理论系统计算了贵金属(金、钯、铂与铑等)原子掺杂在CeO2(111)表面,形成的取代型复合氧化物的结构与电子性质,在此基础上还进一步研究了不同贵金属掺杂催化剂的氧空位形成能,从理论上初步解释了贵金属掺杂型铈基催化剂高活性的原因。
1 模型与计算方法
计算工作是利用基于密度泛函理论的从头算量子力学程序包(Vienna ab-initio simulation package,VASP)完成的[6]。电子交换关联势采用广义梯度近似法(generalized gradient approximation,GGA)中的Perdew-Wang 1991 (PW91)泛函[7]。电子-离子相互作用采用投影缀加平面波赝势(PAW)描述[8],Kohn-Sham单电子态用平面波基组展开并设置截止能为400 eV。布里渊区k点设置为3×4×1[9],自洽场能量收敛标准为1×10-4eV,最大力设置为0.03 eV/Å。
氧空位形成能定义为:EOv=E(Ov/slab)+1/2E(O2)-E(slab)。其中,E(Ov/slab)代表含有氧空位体系的总能量,E(O2)为氧气分子的能量,E(slab)为金属原子掺杂CeO2(111)表面体系的能量。若EOv为大于零,这表明氧空位的形成需要克服一定的能垒,EOv值越小,说明氧空位越容易形成。
2 结果与讨论
2.1MxCe1-xO2模型催化剂的几何结构分析
本文详细的计算了在CeO2(111)表面掺杂Au、Pd、Pt与Rh等不同贵金属原子形成的掺杂模型催化剂结构;在此基础上,优化计算了形成氧空位的模型催化剂,并计算了相应的氧空位形成能。优化的构型如图2所示,相对应的氧空位形成能以及几何参数列于表1。
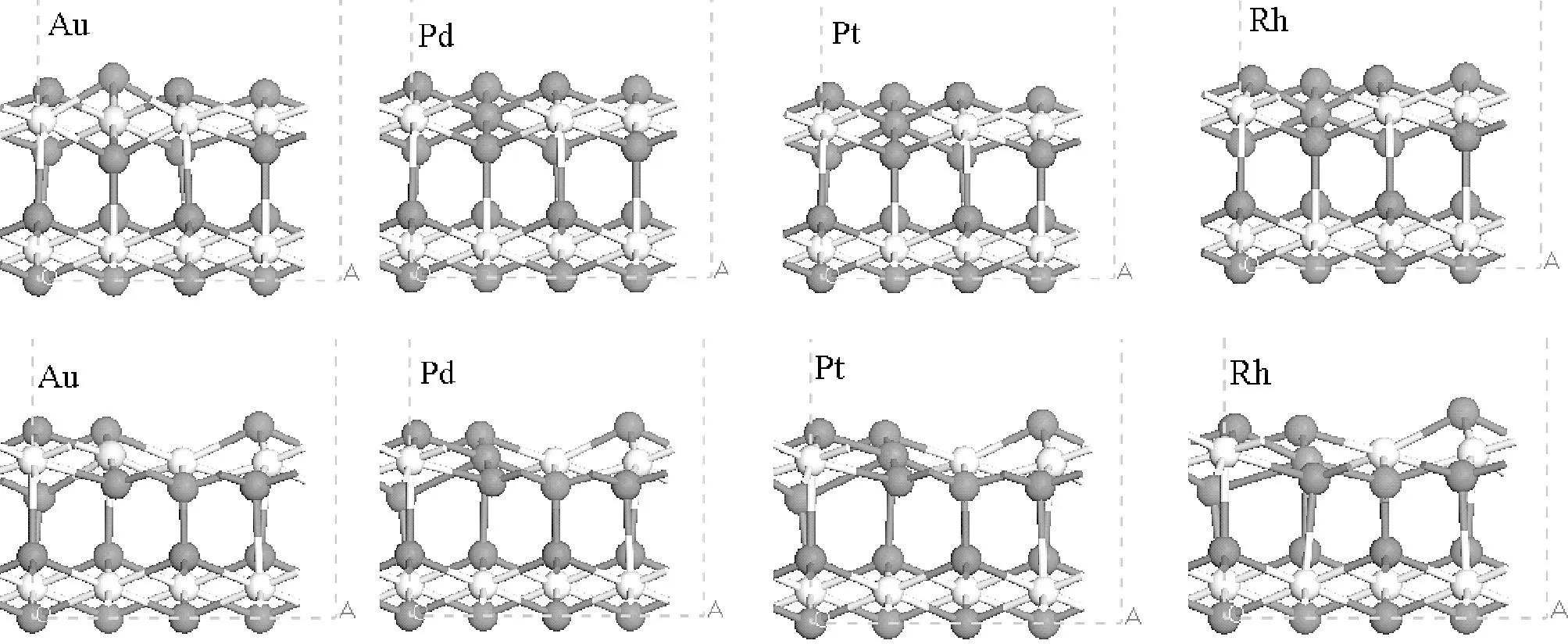
图2 MxCe1-xO2模型催化剂优化构型
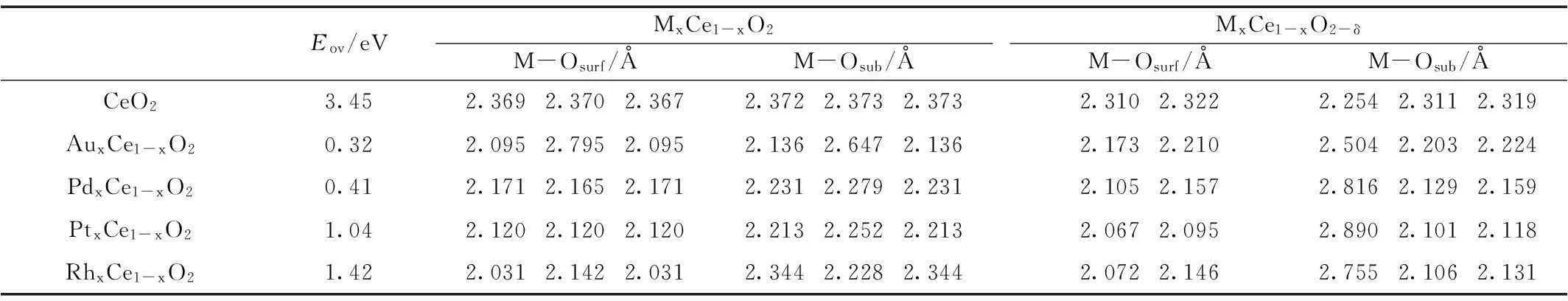
Eov/eVMxCe1-xO2M-Osurf/ÅM-Osub/ÅMxCe1-xO2-δM-Osurf/ÅM-Osub/ÅCeO23.452.3692.3702.3672.3722.3732.3732.3102.3222.2542.3112.319AuxCe1-xO20.322.0952.7952.0952.1362.6472.1362.1732.2102.5042.2032.224PdxCe1-xO20.412.1712.1652.1712.2312.2792.2312.1052.1572.8162.1292.159PtxCe1-xO21.042.1202.1202.1202.2132.2522.2132.0672.0952.8902.1012.118RhxCe1-xO21.422.0312.1422.0312.3442.2282.3442.0722.1462.7552.1062.131
表1详细的列出了不同贵金属原子掺杂CeO2(111)表面的氧空位生成能及相应的M-O键长。蒋仕宇等计算结果证明,在体相CeO2(111)表面氧空位形成能较大,为3.45 eV;当我们掺杂贵金属原子后,氧空位形成能的到大幅降低降低,掺杂Au、Pd、Pt和Rh形成的复合型催化剂表面氧空位形成能分别为0.32、0.41、1.04与1.42 eV,降幅高达为91%、88%、70%与59%。这充分说明贵金属原子的掺杂可以有效的促进氧空位的生成,在此基础上,形成活性氧物种,参与VOCs催化氧化反应,掺杂贵金属的催化剂的催化活性明显高于纯CeO2载体。
几何结构方面,当金属原子取代Ce原子掺杂至CeO2(111)表面后,贵金属原子与周围氧原子之间的距离,即M-O的键长,比Ce-O键的键长略有减小。在化学计量的CeO2(111)中铈氧键的键长约为2.370 Å,掺杂贵金属后形成的复合型催化剂体系的M-O键长均有一定程度的缩短,这可能是由于掺杂的贵金属原子的半径比较铈原子的半径小,因此导致相应的M-O键长比Ce-O键的键长短。如果将M周围同一层的氧原子从左到右依次做记号为1、2、3,那么贵金属掺杂优化后的模型催化剂,不管是表层氧还是次表层氧,M-O1键长与M-O3均相等,而且呈现对称性,而M-O2键表现出一定的特殊性。特别是Au-O2键的键长(2.795、2.647 Å)比其它M-O键偏长,即掺杂Au后的表面氧原子将会向表面伸展较长,因此造成其氧空位比较容易形成,这与Au掺杂体系表面氧空位形成能在四种掺杂金属中最低的计算结果相一致。
当掺杂不同种类贵金属原子形成的MxCe1-xO2模型催化剂在形成氧空位后,因为贵金属原子的引入,使其构型也发生了不同程度的变化。从表1中数据看到,次表层M-O1键长均有一定程度的拉长,Au、Pd、Pt与Rh与Osub的键长分别为2.504、2.816、2.890与2.755 Å,而其它M-O键长都有一定程度的缩短。Nolan等[10]研究表明将Al3+、Sc3+、Y3+和In3+掺杂在CeO2(110)表面结构中,其掺杂离子和氧原子之间的键长也呈现出相似的特征规律。
2.2MxCe1-xO2模型催化剂的电子态密度分析
为了更加详细的了解贵金属原子掺杂CeO2的电子作用机制,本文计算了掺杂前后体系的电子态密度,其结果如图3所示。
图3a为计量的MxCe1-xO2模型催化剂计量表面态密度图,其中虚线代表CeO2(111)表面电子态密度,Ce原子的4f电子特征出现在费米能级以上,O原子的2p电子峰则出现在-4~0 eV,是一个比较宽的宽峰。当贵金属原子掺杂进入二氧化铈后,态密度图总体上依旧表现为CeO2(111)表面的特征峰。但是吸附不同的是,当金属原子掺杂CeO2后,掺杂贵金属的电子峰出现在费米能级附近,这充分说明,掺杂比贵金属吸附相比,掺杂与CeO2(111)表面的相互作用更强,这还可从Bader电荷分析中进一步得到验证。
Rh、Pt与Pd掺杂后形成MxCe1-xO2-δ模型催化剂的电子峰向低能量方向移动,这说明金属原子掺杂使得体系总能量减小,体系趋向更加稳定。与Rh、Pt、Pd三种金属不同,Au掺杂,反而使体系能量变大,说明AuxCe1-xO2-δ模型处在较高的能量状态,因此其表面氧原子比掺杂其他(Rh、Pt、Pd三种金属)形成的模型氧化物更容易失去,从而形成氧空位,这与其氧空位形成能比较低的结果相一致。
图3b为有一个氧空位的MxCe1-xO2模型催化剂计量表面态密度图,其中虚线代表CeO2(111)表面形成氧空位的电子态密度。和氧空位态密度相比而言,金属原子掺杂的氧空位体系电子峰都会向低能量方向移动,而且都在在费米能级附近出现了掺杂金属原子的电子峰。比较计量与氧空位的MxCe1-xO2模型催化剂电子态密度可知,形成氧空位的掺杂体系中Au、Pd、Pt与Rh掺杂峰面积比相对应的无氧空位掺杂体系大。这是因为二氧化铈表面失去一个氧原子形成氧空位后,会留下两个单电子,造成掺杂金属原子失电子数目相对偏少,这一结论还可以从Bader电荷计算中得到验证。


图3 MxCe1-xO2模型催化剂电子态密度图
2.3MxCe1-xO2模型催化剂的Bader电荷分析
为进一步对掺杂金属原子在MxCe1-xO2模型催化剂中电子传递进行分析,本文系统计算了掺杂金属的Bader电荷,并计算了相应贵金属原子的失电子数,列于表2。通过和袁金焕等计算的贵金属原子在CeO2(111)吸附结果相比可以发现,贵金属原子掺杂比吸附行为相比,贵金属原子掺杂失去更多的电子数,这充分证明掺杂金属与表面的相互作用比吸附时更强。这是因为掺杂的贵金属原子进入CeO2体相,掺杂原子与周围的一个铈原子和三个氧原子都有相互作用,这与电子态密度结果相符。另外,掺杂体系贵金属失去的电子数同样表现出了比较好的规律性,依照Au、Pd、Pt与Rh的顺序,掺杂体系(包括形成氧空位的体系)中贵金属原子所失电子逐渐增多,这与不同金属对应的掺杂体系氧空位形成能依次增加相一致。在对掺杂同种金属原子体系的研究发现,在氧空位形成后比形成前金属失去的电子数少,这一规律与电子态密度结果一致。

表2 MxCe1-xO2模型催化剂中贵金属原子的电子数及失电子数
3 结 论
在CeO2(111)中掺杂不同的贵金属原子(Au、Pd、Pt和Rh),使二氧化铈构型发生不同程度的改变,使MxCe1-xO2模型催化剂的氧空位形成能比洁净表面CeO2(111)大幅下降,有效的促进了氧空位的生成,相应掺杂体系的催化活性明显高于体相CeO2载体。贵金属原子掺杂CeO2后的催化剂,在态密度图中的费米能级附近现了相对应的掺杂金属原子的电子特征峰,其与CeO2(111)的相互作用比其在表面吸附更加强烈。依照Au、Pd、Pt与Rh的顺序,掺杂金属原子失电子数目逐步增多,当氧空位在表面形成时,金属原子失电子数比没有氧空位的掺杂表面少。
[1]蒋凯,张秀英,郭崇峰.固体氧化物燃料电池中的电解质[J].稀有金属,2001,25(2):121-125.
[2]洪维民.三效催化剂用CexZr1-xO2:固溶体合成技术与储放氧性能研究进展[J].稀土,2004,25(2):59-64.
[3]Monte R D,Kaspar J.On the role of oxygen storage in three-way catalysis [J].ToP.Catal.,2004,28(1-4):47-57.
[4]袁金焕,滕波涛,赵越,等.贵金属原子在 CeO2(111) 表面吸附的密度泛函理论研究[J].燃料化学学报,2012,40(1):124-128.
[5]袁金焕,滕波涛.金属饰基催化剂上甲醛催化氧化的密度泛函理论研究[D].金华:浙江师范大学化学与生命与科学学院,2011.
[6]Kresse G,Hafner J.Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium [J].Phys.Rev.B.,1994,49(20):14251-14269.
[7]Perdew J P,Burke K,Ernzerhof M.Generalized Gradient Approximation Made Simple [J].Phys.Rev.Lett.,1996,77(18):3865-3868.
[8]Kresse G,Joubert D.From ultrasoft pseudopotentials to the projector augmented-wave method[J].Phys.Rev.B.,1999,59(3):1758-1775.
[9]Monkhorst H J,Pack J D.Special points for Brillouin-zone integrations [J].Phys.Rev.B,1976,13(12):5188-5192.
[10]Nolan M.Charge Compensation and Ce3+Formation in Trivalent doping of the CeO2(110) Surfaee:The Key Role of Dopant Ionic Radius [J].J.Phys.Chem.C,2011,115(14):6671-6681.
Theoretical Study of the Doped CeO2(111) Model by Noble Metals
CUI Peng-fei,JIN Li-fang,ZHANG Jian-wang,YUAN Jin-huan,ZHAO Lei-hong
(Zhejiang Key Laboratory Chemistry on Solid Surfaces,Institute of Physical Chemistry,Zhejiang Normal University,Zhejiang Jinhua 321004,China)
The geometric structures and electronic properties of MxCe1-xO2-δ(111) (M=Au,Pd,Pt or Rh) were systematically studied using density functional theory method.Results suggested that the structures of CeO2(111) surface change due to the doping of noble metal atoms.The surface oxygen vacancy formation energies of Au,Pd,Pt and Rh were 0.32,0.41,1.04,and 1.42 eV,which were much lower than that of the stoichiometric CeO2(111) surface.Therefore,the noble metals doped in ceria can promote the formation of oxygen vacancy,which significantly increased the catalytic activity.The density of state analysis results showed that the doping peaks appeared near the Fermi energy level.The charges of the doped atoms increased as the order of Au,Pd,Pd,and Pt,correspondingly.When forming oxygen vacancy,the charges of doping metal atoms were less than those of MxCe1-xO2without oxygen vacancy.
density functional theory; CeO2; noble metal; density of state; doping
O643
A
1001-9677(2016)011-0028-04
