LCMSMS法测动物源食品中硝呋烯腙残留量的不确定度评定
2016-09-02罗艳霞
罗艳霞
(上海天祥质量技术服务有限公司,上海 200233)
LCMSMS法测动物源食品中硝呋烯腙残留量的不确定度评定
罗艳霞
(上海天祥质量技术服务有限公司,上海200233)
建立了液相色谱-串联质谱测定动物源食品硝呋烯腙的方法,并对其不确定度进行了评定。本文研究了液相色谱-串联质谱测定动物源食品硝呋烯腙含量的方法,并依据CNAS-GL06-2006《化学分析中不确定度的评估指南》和JJF1059.1-2012《测量不确定度评定与表示》规定的原理与方法,分析了实验过程中不确定性的来源,建立了不确定度评定的数学模型,计算并分析了合成不确定度的结果。由结果可知:在液相色谱-串联质谱测定动物源食品硝呋烯腙含量过程中,校准曲线拟合与标准溶液的配置是影响其不确定度的主要因素,液相色谱-串联质谱仪和测量重复性是次要因素。
LCMSMS;动物源性食品;硝呋烯腙;不确定度
我国是畜牧业大国,随着人们对动物源食品由需求型向质量型的转变,动物源食品中的兽药残留已逐渐成为全世界关注的一个焦点[1]。目前在养殖过程中,普遍滥用药物添加剂或高效抗生素等成为影响动物源性食品安全的主要原因。随着人们对食品与环境质量要求的不断提高及对抗生素认识的不断深入,饲料中抗生素的添加所引起的问题愈来愈引起人们的关注。
硝呋烯腙 (Nitrovin)是20世纪70年代中期发现的一种新型抗生素,它是一种呋喃类化学合成药物,它通过增加动物肠粘膜的通透性,从而能有效地提高营养物质在动物消化道中的消化和吸收;还能够有效抑制动物消化道内病原微生物对葡萄糖的利用,使其新陈代谢过程受阻,从而抑制和杀灭多种病原微生物,减少动物感染疾病的机会,提高抗病能力[2]。由于抗生素的耐药性、残留和二重感染的特性,所带来的弊端和危害已成为人类所面临的重大挑战。中华人民共和国农业部国家药品监督管理局公告(第193号):食品动物禁用的兽药及其它化合物清单明确指出硝呋烯腙禁止使用。因此,研究建立动物源性食品中硝呋烯腙的残留检测方法具有重要意义。
目前有关检测动物源性食品中的硝呋烯腙残留量的方法有高效液相色谱法[3],高效液相质谱联用法[4-5]等少量的文献报道,国家也尚未制定相关的检测标准。本文建立了硝呋烯腙的高效液相色谱-串联质谱检测方法,其中硝呋烯腙色谱峰保留时间是3.47min, 线性范围是1.0~50ng/mL,相关系数R为0.999。该方法实验过程简单,有利于食品检测机构的批量检验。并且依据CNAS-GL06-2006《化学分析中不确定度的评估指南》[6]和JJF1059.1-2012《测量不确定度评定与表示》[7]规定的原理与方法,对本文测试方法的准确可靠性进行了详细的分析,用合成不确定度[8-13]为测量结果提供理论基础和科学依据。
1 实 验
1.1材料
1.1.1试剂
硝呋烯腙标准品,Dr.Ehrenstorfer; 乙腈(色谱纯),Fisher;甲醇(色谱纯),Fisher;甲酸(分析纯),国药;二甲基甲酰胺(分析纯),国药。
1.1.2仪器
AB4500型高效液相色谱-串联质谱检测仪,ABSCIEX公司;FE20K型酸度计、MS204S型电子天平、XS105型电子天平,瑞士梅特勒托利多;SF-DL-4M型高速冷冻离心机,上海菲恰尔分析仪器公司;SK250H型超声振荡仪,上海科导超声仪器有限公司。
1.2实验方法
1.2.1标准溶液的配制
精密称取10.00mg硝呋烯腙标准品,置于100mL容量瓶中,加入2mL二甲基甲酰胺溶解,再用甲醇定容,混匀,得到标准储备溶液。用单标线刻度为1mL的移液管准确移取0.1mL标准储备溶液至100mL容量瓶中,甲醇释至刻度,混匀,此标准中间液浓度为 100ng/mL;再用10mL刻度移液管分别移取0.1mL、0.5mL、1.0mL、2.0mL、5.0mL100μg/mL的标准中间液于5只10mL容量瓶中,甲醇释至刻度,混匀,配置成的标准工作液浓度分别为1.0ng/mL、5.0ng/mL、10ng/mL、20ng/mL、50ng/mL(注:此处移液管、容量瓶均为A级)。
1.2.2样品预处理
精密称取5.0000g样品于50mL离心管中,用A级10mL刻度移液管吸取6mL二甲基甲酰胺加入离心管,振摇分散样品,再加入9mL乙腈,超声提取20min,4000r/min冷冻离心10min,过0.45和0.22μm有机膜,供LC-MS-MS分析测定。
1.2.3仪器条件
1.2.3.1液相色谱条件
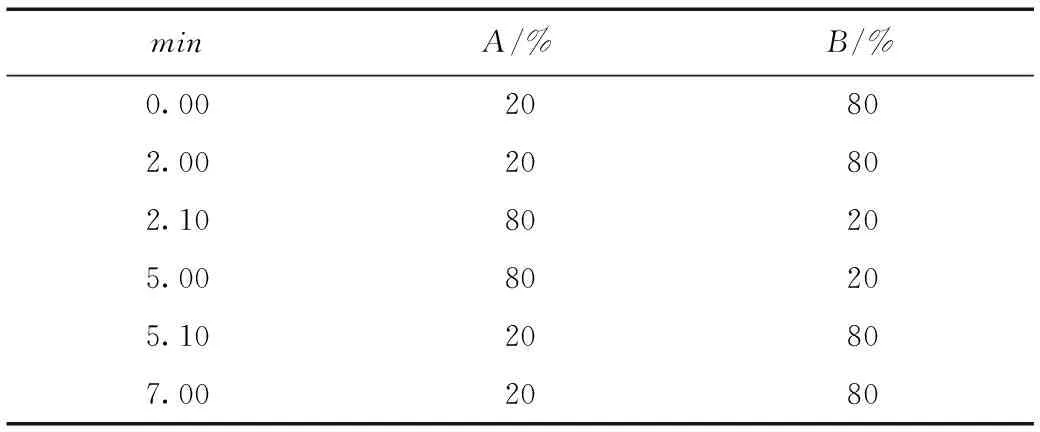
表1 流动相梯度条件Table 1 Mobile phase gradient conditions
色谱条件:色谱柱为AgilentC18(4.6x250mm, 5μm);进样量10μL;流速为0.3mL/min;柱温为 30 ℃;流动相A相甲醇,B相0.1%甲酸水溶液(见表1)。
1.2.3.2质谱条件
离子源:电喷雾(ESI+)离子源;扫描方式:正离子扫描;检测方式:多反应监测(MRM);电压:5500V;雾化气压力:50psi;气帘气压力:35psi;辅助气压力:50psi;碰撞气压力:9psi;离子源温度:500 ℃;碰撞室入口电压:10V;碰撞室出口电压:6V;硝呋烯腙最佳碰撞电压、碰撞能量等如表2所示。

表2 硝呋烯腙的离子对Table 2 The ion pair of nitrovin
1.3数学模型的建立[8-9]
样品中硝呋烯腙含量计算公式如下:
式中: X——样品中硝呋烯腙含量,μg/kg
C——样品溶液中硝呋烯腙浓度,ng/mL
V——样品定容体积,mL
m——样品质量,g
上述计算公式是由测量原理得到的,没有考虑各种随机因素对不确定度的影响,在此引入反映随机影响的重复性系数 frep,其数值等于1。评定不确定度的数学模型应写成如下形式:
1.4测量不确定度因素[8-13]
依据检测过程和数学模型,不确定度的主要因素有:实验重复性条件产生的相对标准不确定度uref(frep);样品称样量产生的相对标准不确定度uref(m);样品定容体积引起的相对标准不确定度uref(V);标准溶液配制引起的相对标准不确定度uref(C);校准曲线拟合引起的相对标准不确定度uref(Co); 液相色谱-串联质谱仪定量重复性产生的相对标准不确定uref(LCMSMS)。合成相对标准不确定度uref(X)计箅公式如下:
2 结果与讨论
2.1实验重复条件下产生的相对标准不确定度uref(frep)
对样品进行7次重复测定所得到数据如表3所示。

表3 样品测定结果Table 3 Samples determination results
重复测量7次的实验标准偏差为:
日常测量时为平行测定,结果是平均值,故平均值相对标准不确定度为:
实验重复条件下产生的标准偏差,考虑了所有的随机影响。因此在分析之后的各因素时,无需考虑随机影响。
2.2样品称样量的相对标准不确定度uref(m)
用天平称取5.0000g样品时,因天平所允许的最大误差为±0.0001g, 按矩形分布分析,称样时天平引入的相对标准不确定度为:
2.3样品定容体积的标准不确定度uref(V)
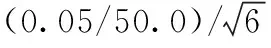
2.4硝呋烯腙标准溶液的相对标准不确度uref(C)
2.4.1配制标准储备液所引入的相对标准不确定度uref(C1)
(1)由硝呋烯腙标准品证书可知其纯度为98.5%,由标准品纯度引入的不确定度为:
(2) 准确称取标准品硝呋烯腙时,天平所允许的最大误差为±0.01mg, 按矩形分布分析,天平引入的相对标准不确定度为:
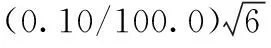
(4)因此配制标准储备液时所引入的相对标准不确定度uref(C1)为:
2.4.2配制标准中间液所引入的相对标准不确定度uref(C2)
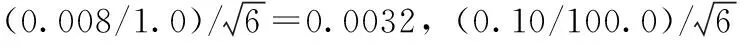
2.4.3配制标准工作液所引入的相对标准不确定度uref(C3)
依据JJG196-2006[14]常用玻璃量器检定规程,据三角形分布分析,配制标准工作液所引入的相对标准不确定度uref(C3)见表4[9]。
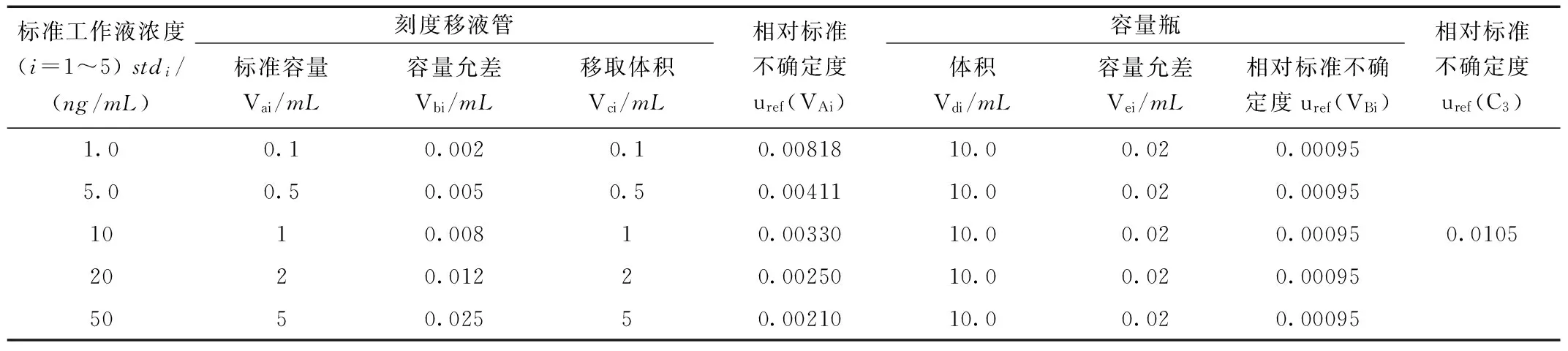
表4 标准工作液配制引入的相对标准不确定度Table 4 The introduction of relative standard uncertainty of standard working liquid
①按三角分布分析,研讨溶液温度与校正时温度差引起的体积不确定度0.00048,可知uref(VAi)为:
②按三角分布分析,研讨溶液温度与校正时温度差引起的体积不确定度0.00048,可知uref(VBi)为:
③综合以上得到相对标准不确定度uref(C3):
2.4.4配制硝呋烯腙标准溶液的相对标准不确定度uref(C)
2.5校准曲线拟合引入的相对标准不确定度uref(Co)
用LCMSMS测定硝呋烯腙标准溶液5个浓度水平,可知相应的峰面积,运用最小二乘法拟合,拟合方程Y=aC+b和相关系数r(见表5)。

表5 测定及由拟合方程计算结果Table 5 Measurement and calculation results by fitting equations
注:a=35242,b=2406,r=0.999。
依据拟合的标准曲线,将样品测定10次,得到的样品溶液浓度测定结果表6所示。
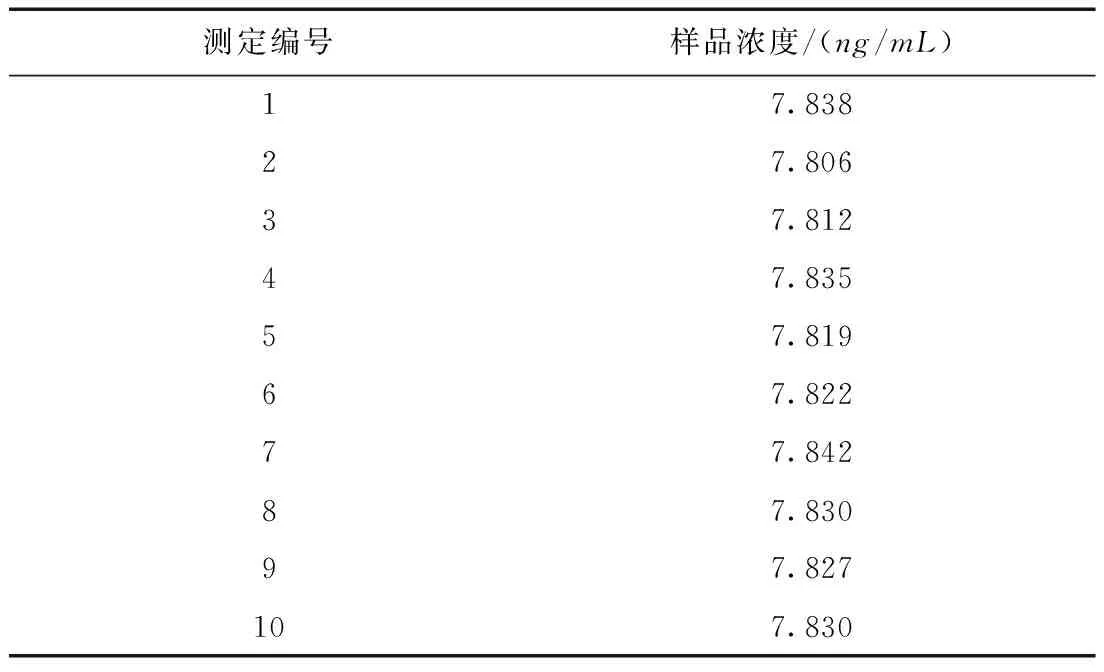
表6 样品溶液浓度测定结果Table 6 Sample solution concentration determination results
计算可知:C0=7.826ng/mL,X=23.48μg/kg
由标准曲线求浓度时的相对标准不确定度uref(C0)由下式计算[9]:
式中:a——标准工作曲线的斜率
P——重复测定样品的数量
m——重复测定标准溶液的数量
n——配制的标准溶液数量
Sy/c——工作曲线的标准偏差
Sc/c——工作曲线各浓度点的离均差平方和
Yij——工作曲线各浓度点的测定响应值
Yi——由线性方程计算所得响应值
Ci——工作曲线各浓度点

C0——测试样平均浓度
由以上公式计算的结果见表7。
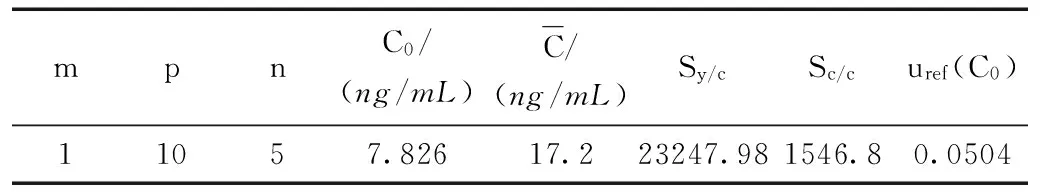
表7 最小二乘法拟合标准曲线时引入的不确定度uref(C0)Table 7 Least squares fitting in uncertainty of standard curve
2.6液相色谱-串联质谱在测量过程中所引入的相对标准不确定度uref(LCMSMS)
2.7合成相对标准不确定度及评定扩展不确定度
2.7.1合成相对标准不确定度
依据文中各项不确定度分量(见表8),合成得到相对标准不确定度uref(X):
=0.0548

表8 各相对标准不确定度分量汇总表Table 8 Each relative standard uncertainty componentssummary table
2.7.2扩展不确定度
当包含因子k=2,置信水平约为95%,依据公式计算可得相对扩展不确定度uref(X):
Uref(X)=uref(X)×k=0.1096
依据表6计算得,动物源性样品中硝呋烯腙测定的残留量的最佳预估值为23.48μg/kg,故其扩展不确定度U为:
U=23.48×Uref(X)=2.6μg/kg
3 结 论
本实验采用液相色谱-串联质谱法测定动物源性样品中硝呋烯腙残留量,当k=2,置信水平是95%时,测得的硝呋烯腙含量为(23.48±2.6)μg/kg。本文中实验过程简单易行,用来对动物源性食品中硝呋烯腙的测定准确可靠性较高。
对实验过程及不确定度分量结果综合分析可知:在液相色谱-串联质谱测定动物源食品硝呋烯腙残留量过程中,校准曲线拟合与标准溶液的配置是影响其不确定度的主要因素,液相色谱-串联质谱仪和测量重复性是次要因素。
[1]郭晓娟.兽药残留产生的原因及危害[J].养殖技术顾问,2011,5(03):12-14.
[2]陈永丰,印遇龙,谭支良,等.硝呋烯腙对断奶仔猪生产性能的影响[J].中国畜牧杂志,2003,39(03):29-30.
[3]LinSY,JengSL.High-performanceliquidchromatographicdeterminationofcarbadox,olaquindox,furazolidone,nitrofurazone,andnitrovininfeed[J].JournalofFoodProtection,2001,64(8):1231-1234.
[4]王金荣,李德发,张丽英,等.硝呋烯腙的纯化及其在饲料中的HPLC及LC-MS检测技术研究[J].分析测试学报,2006,26(z1):99-100.
[5]徐彦辉,何俊,顾亮,等. 液相色谱串联质谱法测定动物源性食品中硝呋烯腙的残留量[J].化学分析计量,2011,20(4):29-32.
[6]中国合格评定国家认可委员会,CNAS-GL06:2006化学分析中不确定度的评估指南[S].北京:中国计量出版社,2006.
[7]国家质量技术监督局.JJF1059.1-2012测量不确定度评定与表示[S].北京:中国计量出版社,2012.
[8]汤海靑,顾晓俊,殷居易,等.高效液相色谱法测定葡萄酒中苯甲酸含量的不确定度评定[J].食品工业科技,2013,34(15):295-298.
[9]烟利亚.高效液相色谱法测定淀粉中马来酸含量的不确定度评定[J].广州化工, 2014,42(20):145-148.
[10]蔡秋,朱明.高效液相色谱法测定茶饮料中苯甲酸结果不确定度评定[J].食品科学,2007(02):277-280.
[11]汪辉,曹小彦,彭新凯,等.高效液相色谱法测定小麦粉与大米粉中甲醛次硫酸氢钠含量的不确定度评定[J].食品科学,2009,30(12):205-208.
[12]杨洋,徐春祥,车文军.高效液相色谱法测定奶粉中的三聚氰胺及其不确定度分析[J].食品科学,2010,31(04):250-253.
[13]朱佳,孙杰.液相色谱法测定果汁中糖精钠的不确定度研究[J].食品研究与开发, 2012,33(02):156-158.
[14]国家质量监督检验检疫总局,JJG196-2006常用玻璃量器检定规程[S].北京:中过国计量出版社,2006.
Uncertainty Evaluation for the Determination of Nitrovin Residues inAnimal-originFoodbyLiquidChromatographyTandemMassSpectrometry
LUO Yan-xia
(Shanghai Intertek Testing Services Co., Ltd., Shanghai 200233, China)
AliquidchromatographytandemmassspectrometrydeterminationofNitrovinresiduesinanimal-originfoodwasestablished,andthemeasurementuncertaintyofthismethodwasevaluated.Theliquidchromatography-tandemmassspectrometrydeterminationofanimaloriginfoodmethodofnitrovincontentwasstudied,accordingtotheprincipleandmethodinCNAS-GL06-2006GuidanceonEvaluatingtheUncertaintyinChemicalAnalysisandJJF1059.1-2012EvaluationandExpressionofUncertaintyinMeasurement,thesourceofuncertaintyintheexperimentprocesswasanalyzed,themathematicalmodelofuncertaintyevaluationwasestablished,theresultofthesynthesisofuncertaintywascalculatedandanalyzed.Theresultsshowedthatinthedeterminationofanimaloriginfoodofnitrovincontentintheprocess,calibrationcurvefittingandtheconfigurationofthestandardsolutionwerethemainfactorsaffectingtheuncertainty,liquidchromatography-tandemmassspectrometerandrepeatabilityweresecondaryfactors.
LCMSMS;animal-originfood;nitrovin;uncertainty
罗艳霞,女,本科,主要从事食品检测工作。
O657.7+2
A
1001-9677(2016)013-0136-05
