莫格列扎中间体的合成*
2016-09-02朱高峰范钰新王建塔
朱高峰,夏 晶,曹 俊,范钰新,王建塔,汤 磊
(贵州医科大学药学院,贵州 贵阳 550004)
莫格列扎中间体的合成*
朱高峰,夏晶,曹俊,范钰新,王建塔,汤磊
(贵州医科大学药学院,贵州贵阳550004)
设计与合成阿格列扎中间体2-(5-甲基-2-苯基-4-噁唑基)乙基甲烷磺酸盐。以L-天冬氨酸为原料先与甲醇和氯化亚砜反应生成相应的甲酯盐酸盐(15),化合物15先后发生亲核加成、环合、水解和亲核取代反应得到目标物(化合物1)。合成的化合物及中间体均经过核磁共振氢谱进行了结构表征确证结构与目标产物一致。该合成方法可靠,质量可控,能用于2-(5-甲基-2-苯基-4-噁唑基)乙基甲烷磺酸盐的合成。
莫格列扎;中间体;合成
2-(5-甲基-2-苯基-4-噁唑基)乙基甲烷磺酸盐,为阿格列扎、替格列汀、莫格列扎等口降糖药的重要中间体。该中间体在本课题组所合成的GY3系列化合物中,在体内外均具有很好的降糖活性。目前该化合物的合成方法主要有如下三种。
方法一(图1)[1]:原料苯甲醛(2)与2,3-丁二酮-1-肟钠(3)在酸性条件下发生席夫碱反应,后在三氯氧磷中发生氯代反应生成化合物5,后者与氰化钠反应后生成带有氰基的化合物6,化合物6先后与氢氧化钾和甲磺酰氯反应生成目标产物化合物1。总收率约50%。
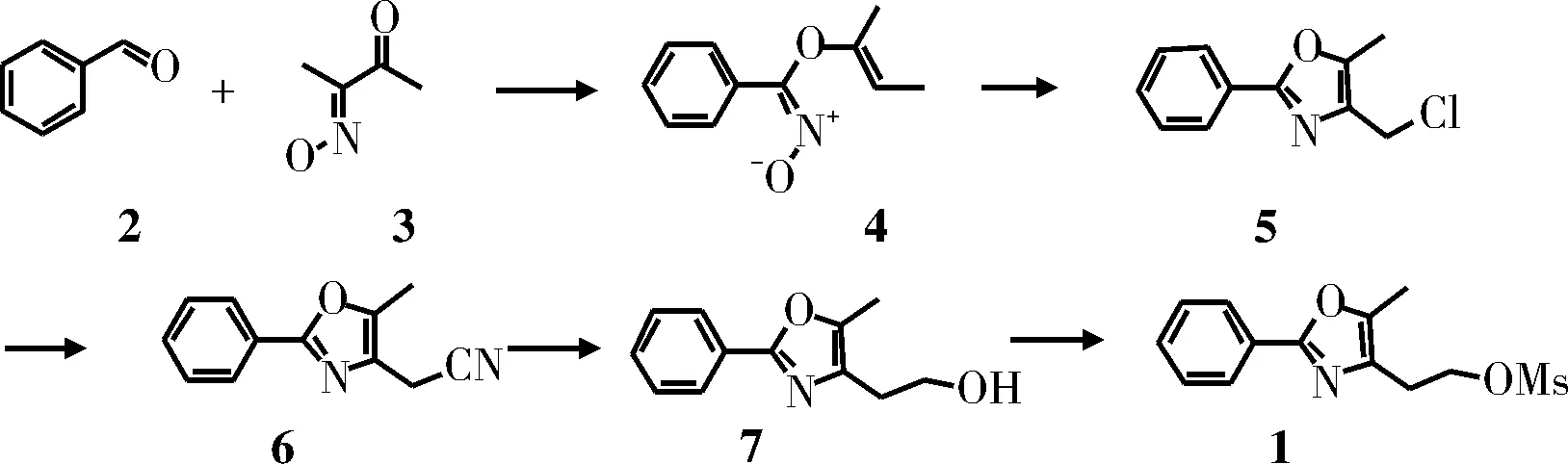
图1 方法一
方法二(图2)[2-3]: 4-溴代-3-羰基-戊酸甲酯与本甲酰胺在甲苯中回流,发生亲核取代和环合反应生成中间体10,后者水解后与甲磺酰氯反应生成目标产物1。总收率31%。
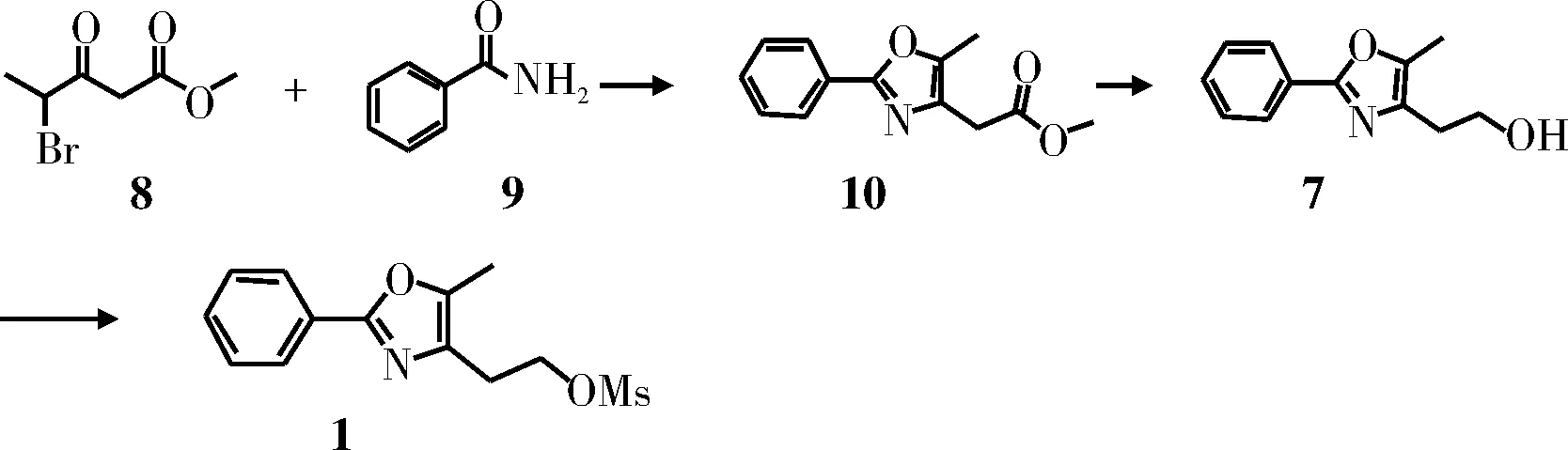
图2 方法二
方法三[4]:天冬氨酸(12)与苯甲酰氯反应生成中间体13;后者经Dakin-West反应后,在酸性条件下脱水环合得到中间体15;中间体15经还原剂还原生成醇,最后与甲酸酰氯反应,得到目标产物1。收率未报道。
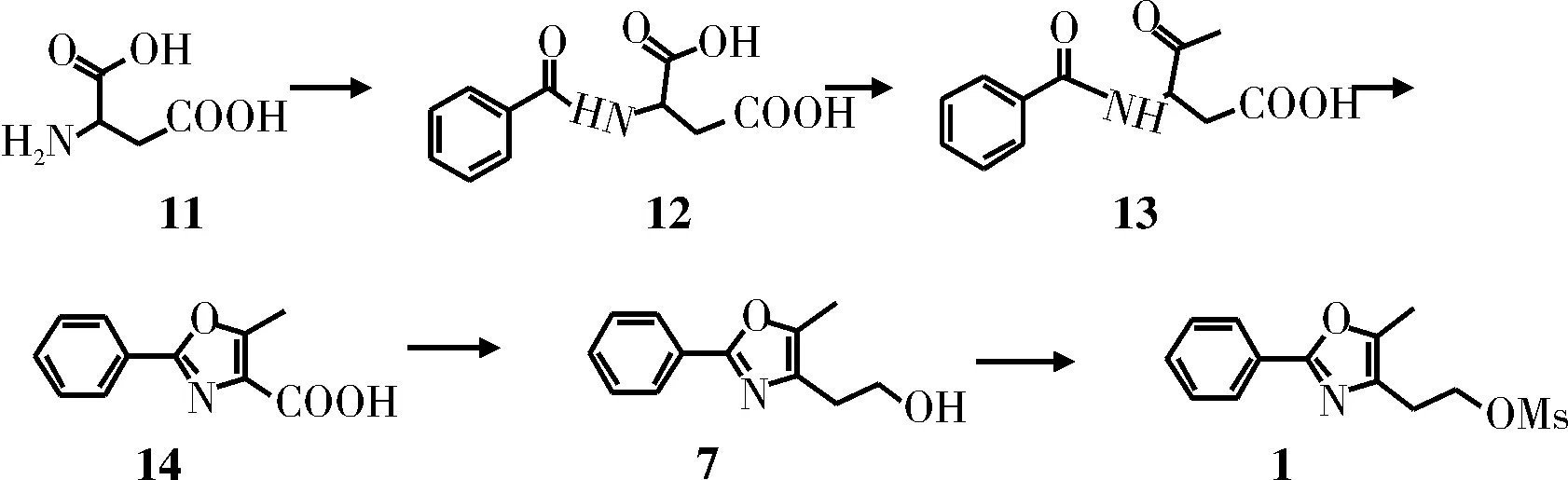
图3 方法三
通过对上述三种合成方法比较发现:方法一收率相对较高,但是原料2,3-丁二酮-1-肟钠市场不易购买、氯代三氯氧磷刺激下较大,且用到了毒性较大的氰化钠,对合成操作者和环境都可能会造成很大危害。方法二收率相对较低、合成路线短,但是原料4-溴代-3-羰基-戊酸甲酯在市场上仍不易购买。路线三合成原料苯甲酰氯稍有刺激性,反应比较温和。
在参考上述三种路线的基础上,以L-天冬氨酸为原料先与甲醇和氯化亚砜反应生成相应的甲酯盐酸盐(15),化合物15先后发生亲核加成、环合、水解和亲核取代反应得到目标物(化合物1,方法四)。

图4 方法四
1 材料和方法
Mercury 400 核磁共振仪,美国Varian公司;Tektronix X-4熔点仪(未校正),北京泰克。
L-天冬氨酸,国药集团;苯甲酰氯,阿拉丁试剂;其余原料及试剂均为分析纯。
2 合成路线和方法
2.1L-天冬氨酸-4-甲酯盐酸盐(15)的合成
640 mL甲醇冰盐浴冷至-10 ℃左右,滴加60 mL(0.8 mol)氯化亚砜,滴加时控制内温小于-5 ℃。滴毕,加入80 g(0.6 mol)L-天冬氨酸。室温反应90 min,缓慢加入乙酸乙酯4.2 L,搅拌60 min,冰箱冷藏放置60 min,抽滤。滤饼减压干燥得112 g白色固体(15),收率92%。mp:188~190 ℃(文献值[5]:190 ℃)。
2.2N-苯甲酰基-L-天门冬氨酸-4-甲酯(16)的合成
化合物15 79 g加水347 mL溶解,冰水浴冷却,加入Na2CO3水溶液(91.3 g溶于450 mL水)。搅拌10 min后,加入苯甲酰氯52.7 mL,5 ℃搅拌90 min,室温搅拌2 h。加水500 mL,用二氯甲烷231 mL 洗涤水相。浓盐酸调水相pH 至2,用乙酸乙酯600 mL萃取,水相再次用乙酸乙酯300 mL萃取。合并有机相,分别用水、饱和食盐水各300 mL洗涤一次,无水硫酸镁干燥,浓缩至约500 mL,放置析晶。抽滤,滤饼减压干燥得95.4 g白色固体(16),收率88%。mp:116~119 ℃(文献值[6]:118~119 ℃)。lH NMR (CDCl3), δ(ppm): 7. 84~7. 82 (m,2H), 7.44~7. 42 (m, 3H), 5. 16~5. 11 (m, 1H), 3.70 (s, 3H), 3.23 (dd,J=4 Hz, 16 Hz, 1H), 3.08 (dd,J=4 Hz, 16 Hz, 1H)。
2.35-甲基-2-苯基恶唑-4-乙酸甲酯(10)的合成
化合物16 33.1 g、 乙酸酐110 mL、 浓硫酸3.8 mL,混合液于90 ℃反应60 min。冷却至室温,加水120 mL稀释,二氯甲烷100 mL萃取。有机相分别用水、10%碳酸钠、饱和食盐水洗涤,无水硫酸钠干燥。最后减压浓缩得21 g,收率68.9%。1H NMR (CDCl3), δ (ppm): 7. 94~7. 91 (m, 2H), 7. 46~7. 38 (m, 3H), 3.71 (s, 3H), 3.49 (s, 2H), 2.36 (s, 3H)。
2.42-(5-甲基-2-苯基-4-噁唑基)-1-乙醇(7)的合成
化合物10 20 g溶于110 mL 四氢呋喃溶液中,冰水浴下滴入3.4 g 四氢锂铝溶于90 mL四氢呋喃的混悬液。滴毕,室温反应20 h,滴加水4 mL淬灭反应。加入15% NaOH(8 mL)溶液,搅拌30 min,抽滤,滤液蒸干。向剩余物中加入乙酸乙酯、混匀,过滤除去不溶物,滤液放置析晶。抽滤、减压干燥,得白色固体19.5 g(7),收率90%。 mp: 65~67 ℃(文献值[7]: 66~67.7 ℃)。1H NMR (CDCl3), δ(ppm):7.98~7.76 (m, 2H), 7.44~7.42 (m, 3H), 3.93 (t, 2H), 2.71 (t, 2H), 2.30 (s, 3H)。
2.52-(5-甲基-2-苯基-4-噁唑基)乙基甲烷磺酸盐(1)
化合物7 15 g,溶于70 mL二氯甲烷中,冰水浴下加入甲磺酰氯12 mL、三乙胺24 mL、滴加入此化合物中,室温反应20 h。反应毕,反应液分别用稀盐酸、饱和碳酸氢钠、饱和食盐水洗,无水硫酸钠干燥。蒸除溶剂后,用甲基叔丁基醚重结晶,得白色固体(1)17.8 g,收率86%。mp:85~87 ℃(文献值[2]: 86~88 ℃);1H NMR (CDCl3), δ (ppm): 8.15~7.86 (m, 2H), 7.55~7.38 (m, 3H), 4.48(t,J=7.0 Hz, 2H), 2.98 (t,J=7.0 Hz, 2H), 2.94 (s, 3H), 2.50 (s, 3H)。
3 结果与讨论
3.1化合物15的制备
制备中间体化合物15时,由于靠近氨基位置羧基的位阻大、并且只加入一当量的氯化亚砜量的原因,该反应只在远离氨基的羧基上形成甲酯。若提高反应的温度或提高氯化亚砜的用量,反应就会生成双甲酯化产物。
3.2化合物16的制备
制备中间体化合物16时,可考虑反应在非水环境中进行;这样可以减少苯甲酰氯在水中变质,并提高此步反应的收率。此步反应,加入碳酸氢钠的目的是为了与反应生成的副产物氯化氢反应,使反应向生成物方向进行。也可用三乙胺、N-甲级吗啉等 有机碱代替碳酸氢钠,
3.3化合物7的制备
制备化合物7时,加水是为了淬灭反应。四氢锂铝转变为氢氧化锂和氢氧化铝,为胶体状沉淀,会包裹生成的产物,同时抽滤困难。需加入氢氧化钠溶液,使胶体状物分散开,萃取方便,提高反应的收率。
3.4结果
反应总收率43.1%,合成的化合物及中间体均经过核磁共振氢谱进行了结构表征确证结构与目标产物一致。该合成方法可靠,质量可控,能用于2-(5-甲基-2-苯基-4-噁唑基)乙基甲烷磺酸盐的合成。
[1]Hee Oon Han, Seung Hae Kim, Kyoung-Hee Kim, et al. Design and synthesis of oxime ethers of a-acyl-b-phenylpropanoicacids as PPAR dual agonists[J]. Bioorganic & Medicinal Chemistry Letters, 2007, 17:937-941.
[2]Jingbao Liu, Faqin Jiang, Yan Jin, et al. Design, synthesis, and evaluation of 2-substituted ethenesulfonic acid ester derivatives as protein tyrosine phosphatase 1B inhibitors[J]. European Journal of Medicinal Chemistry, 2012, 57: 10-20.
[3]杨玉社,俞娟红,李站,等.一类新的苯并杂环类化合物、其制备方法和用途[P]. CN101092415A.
[4]Delmedico, Mary Katherine. Methods of treating or preventing cognitive impairment using indaneacetic acidderivatives and their preparation[P].WO 2010/141696A1.
[5]D. COLEMA. Synthtic Polypeptides. Part I V. Preparation and Polymerisation of Oxaxolid-2: 5-diones derived from a-Amino-dicurboxylic Acids[J]. Journal of the Chemical Society, 1951: 2294-2295.
[6]Wieczorek Wanda, Bukowska-Strzyzewska Maria, Olma Aleksandra. α-Hydroxym-ethylaspartic acid: Synthesis and absolute configuration by X-ray analysis of its derivative (+)-4-benzoylamino-4-carboxy-γ-butyrolactone[J]. Journal of Crystallographic and Spectroscopic ResearchApril, 1991, 21(2):107-112.
[7]Wachenfeldt Henrik V, Paulsen Filip, Sundin Anders, et al. Synthesis of Substituted Oxazoles from N-Benzyl Propargyl Amines and Acid Chlorides[J]. European Journal of Organic Chemistry, 2013, 21: 4578-4585.
Synthesis of Muraglitazar Intermidate*
ZHU Gao-feng, XIA Jing, CAO Jun, FAN Yu-xin, WANG Jian-ta, TANG Lei
(College of Pharmacy, Guizhou Medical University, Guizhou Guiyang 550004, China)
2-(5-methyl-2-phenyloxazol-4-yl)ethyl methanesulfonate(1), a key intermediate for the preparation of dual PPARα/PPARγ agonist Aleglitazar, was synthesized. Starting from L-aspartic acid, the target compound was obtained via 5 successive steps, including esterification, nucleophilic addition, cyclization, hydrolization and nucleophilic substitution. All the desired compounds were identified by1H NMR, and were consistent with the chemical structures. The synthetic procedure was simple, controllable, and suitable for scale-up synthesis of 2-(5-methyl-2-phenyloxazol-4-yl)ethyl methanesulfonate.
muraglitazar; intermidate; synthesis
贵州省普通高等学校药物化学工程研究中心(黔教合KY字[2014]219号);贵州省科技厅基金(黔科合J字[2012]2034号)。
朱高峰(1982-),男,实验师,主要从事药物开发和应用。
汤磊(1974-),男,教授,主要从事药物开发和应用。
TQ914.2
B
1001-9677(2016)013-0082-03
