半再生重整分子水平反应动力学模型
2016-08-11王杰广BENNETCraing马爱增KLEINMichael郭锦标
周 祥, 王杰广, 侯 震, BENNET Craing A,马爱增, KLEIN Michael T, 郭锦标
(1.中国石化 石油化工科学研究院, 北京 100083; 2.Energy Institute, University of Delaware, Newark, USA)
半再生重整分子水平反应动力学模型
周祥1, 王杰广1, 侯震2, BENNET Craing A2,马爱增1, KLEIN Michael T2, 郭锦标1
(1.中国石化 石油化工科学研究院, 北京 100083; 2.Energy Institute, University of Delaware, Newark, USA)
摘要:根据实验数据建立了半再生重整分子水平反应动力学模型。模型包括305个C1~C12分子组分及864个化学反应,其中包括数十种烯烃及C6~C9所有芳烃异构体的生成与转化。以Langmuir-Hinshelwood- Hougen-Watson(LHHW)方程模拟反应速率,采用线性自由能关联方法(LFER)获取动力学参数,利用实验数据对模型进行了校正及检验。结果表明,芳烃脱烷基速率较低,应区分于加氢裂化反应;芳烃甲基迁移速率不容忽视,对C8+芳烃产物分布有影响;模型对贫、富芳烃2种原料在10组不同反应条件下的C5+液收、正构烷烃和环烷烃产率的预测较为准确,对C6~C9芳烃组分产率(质量分数)的预测偏差平均值小于3%。
关键词:分子水平; 半再生重整; 动力学模型; 固定床
催化重整可将石脑油中的环烷烃和链烷烃转化为芳烃,以生产高辛烷值汽油调合组分或芳烃,还可为加氢装置提供氢气,是石油炼制的主要工艺过程之一。开展分子水平催化重整反应动力学研究有助于加深对其转化过程的认识,不仅可实现重整产物分布和产品性质的预测,还可为催化剂性能评价、装置操作优化乃至炼厂物流优化提供支持。
1959年Smith[1]提出的4集总模型是最早的催化重整反应动力学模型,而后Froment[2]、Ramage等[3]和周红军等[4]开发了不同复杂度的集总水平模型。这些模型多依据PINA族和碳数将组分划分为数个至数十个集总,并以同等数量级的集总转化关系模拟体系中的化学反应。本质而言,催化重整体系一般包含数百个C1~C12组分,近千个化学反应,集总水平模型所能体现的化学信息与之尚存在较大差距,其动力学参数所能表达的定量信息也较为有限,适用性受到一定制约。20世纪90年代以来,随着分析技术的发展以及催化重整反应化学认识的深化,测定重整体系的分子组成并研究其反应转化过程具备了可行性。Antos等[5]提出从分子水平模拟芳烃组分的生成,Joshi等[6]初步建立了C1~C10催化重整分子水平反应动力学模型,其中包含79个组分及480个化学反应。目前,在组分和反应两方面都接近实际规模的催化重整反应动力学模型较少。笔者针对半再生重整建立了分子水平反应动力学模型,利用实验数据对动力学参数进行了校正,并验证了模型对主要产物分布的预测效果。
1 实验部分
1.1原料
采用取自炼化企业的N+A芳潜值分别为质量分数52%和32%的2种加氢精制石脑油作为半再生重整的贫、富芳烃原料。
1.2实验方法
采用中国石化石油化工科学研究院(石科院)自行设计建立的固定床单反应器装置和石科院开发的某铂铼半再生重整催化剂,在反应压力1.0 MPa和1.2 MPa,氢/油体积比1000,反应温度470~510℃,空速1.0~2.0 h-1的条件下进行实验。
共完成18组实验,每组实验的反应器出口产物经冷却、分离后收集液体样品。采用气相色谱仪分析原料和液体产物组成。
1.3实验数据
气相色谱分析结果显示,原料和液体产物中包含C2~C12石油烃组分,其中C2~C8组分、C9链烷烃、部分C9环烷烃、C9芳烃以及C10~C12正构烷烃可较清晰分割为单体,其余C9环烷烃和C10~C12组分相互重叠,表现为“混合峰”,仅能体现其中所含组分的碳数和沸点范围。因此共检测出200余种单体烃组分并测得其含量,其余组分包含于数十个“混合峰”中。值得注意的是,在解析出的单体烃中包含数十种C4~C8烯烃组分。
2 半再生重整反应网络构建
2.1反应类型
对于催化重整反应化学已有较全面的认识,金属中心与酸中心双功能催化反应机理得到了较广泛认同[7]。在金属中心作用下发生六元环烷烃脱氢(Aromatization)、氢解(Hydrogenolysis)等反应,而更多反应类型则需双中心协同作用,包括五元环烷烃扩环(Naphthene ring expansion)、链烷烃异构(Paraffin isomerization)、链烷烃脱氢环化(Paraffin dehydrocyclization)、加氢裂化(Hydrocracking)等。笔者构建的反应网络中包括上述主要反应类型,同时考虑了以下因素。
(1)除加氢裂化和氢解外,其余反应类型均视为可逆反应。
(2)重整产物中有少量烯烃组分,因此设定链烷烃和环烷烃脱氢反应类型,以描述烯烃组分的生成,并视为可逆反应。
(3)以五元环烷烃为链烷烃脱氢环化直接产物。
(4)在催化重整体系中,芳烃脱烷基(Aromatic dealkylation)反应机理较为复杂,有别于芳烃侧链和链烷烃的加氢裂化,因此将其单独设为一个反应类型。
(5)在酸中心作用下,芳烃组分的甲基侧链有可能向邻位迁移,即发生芳烃甲基迁移(Aromatic methyl shift)反应,为准确地计算芳烃组分产率,在反应网络中考虑此类反应。
(6)一般认为高碳数环烯烃,尤其是五元环二烯烃是生焦前驱物[7-8],但对于生焦反应机理尚无明确结论,因此以五元环二烯烃聚合反应简化描述生焦反应。
2.2反应网络
催化重整体系包含C1~C12石油烃组分,其中可能出现的异构体数以千计,受分析水平和计算能力所限,在反应网络中全面考虑所有可能组分并不现实。笔者对比2种原料和18个液体产物的单体烃分析结果,选取出现频率及含量较高的组分,重点考察C4~C9组分,归纳出具有代表性的分子结构。对于未能明确解析的C10~C12组分,参考归纳出的分子结构和反应化学特性,以数个模型化合物为代表,如图1所示,其中—R表示直链取代基。
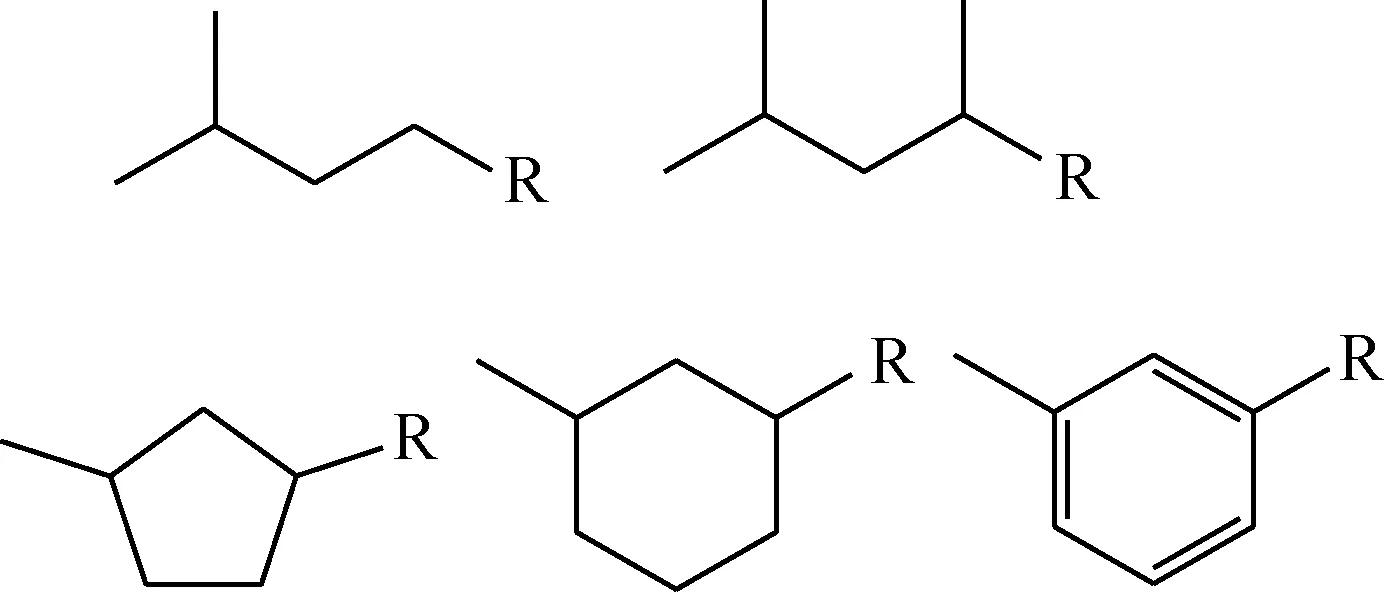
图1 C10~C12模型化合物Fig.1 Model compounds for C10-C12 species
对于归纳及设定的分子组分,基于上述反应类型,判断其在重整体系中可能发生的化学反应。考虑到动力学模型计算的复杂度,对反应进行了以下简化。
(1)在金属中心作用下,链烷烃和烷基侧链上的C—C键均可能发生氢解断裂,鉴于反应物数量较多,限定氢解仅发生于末端2个C—C键,甲烷和乙烷作为其标志产物。
(2)链烷烃异构体较多,其异构化可能存在环丙烷和甲基迁移等多种机理[8],因此体系中可能发生的链烷烃异构化反应数量庞大,参考分析结果中主要异构烷烃的结构类型,约束此类反应不生成支链度过高的产物。
(3)C10~C12组分未被明确解析,限定链烷烃模型化合物和环烷烃、芳烃模型化合物的侧链不发生异构化反应,芳烃模型化合物不发生甲基迁移反应。
最终形成的反应网络包含305个组分,其中约100个组分为中间产物,以及864个化学反应。表1列出了C4~C9异构烷烃、烯烃、环烷烃及芳烃的组分数,各主要反应类型对应的反应数量列于表2。需要说明的是,分析结果提供了9种C9芳烃的完整解析,其中包括茚满。重整体系中的双环产物较为特殊,Klein等[8]认为,双环化合物由五元环烯烃组分经过Diels-Alder反应生成。笔者参考其思路以式(1)所示反应路径表达茚满的生成。


(1)

表2 催化重整各主要反应类型对应的反应数
3 半再生重整反应动力学参数获取
3.1反应速率方程
催化重整是气-固两相反应体系,化学反应机理包括吸附、表面反应和脱附3个步骤,笔者以沿用度较高的LHHW方程[9]模拟反应速率。对于固定床反应器,假设物料以平推流形式与催化剂接触,反应速率以反应物沿床层轴向的消耗量表达。体系中大量存在“X↔Y+Z”型单分子反应,若表面反应为速控步骤,则其LHHW速率方程可由式(2)表示。式(2)中,k为表面反应速率常数,s-1;KX为组分X在活性中心的吸附平衡常数,kPa;pX为组分X的气相分压,kPa;S0为催化剂表面活性中心的浓度,mol/L;K为反应平衡常数,kPa;I表示其它吸附于活性中心的组分。
(2)
3.2动力学参数
由式(2)可见,为实现定量计算,表面反应速率常数、体系中各组分在金属中心和酸中心的吸附平衡常数,以及反应平衡常数是重要的参数。对于吸附平衡常数,笔者研究了不同烷烃、环烷烃和芳烃模型化合物在类似体系中的吸附平衡情况,并将其吸附平衡常数与分子结构进行关联,如式(3)、式(4)所示。式(3)、式(4)中,NAR,X、NNR,X和NSC,X分别为组分X分子结构中芳环、环烷环和饱和碳链的碳数;m、nAR、nNR和nSC为关联参数,针对催化剂确定这些参数即可计算各组分吸附平衡常数。
lnKX=m+NX/RT
(3)
NX=f(nAR·NAR,X,nNR·NNR,X,nSC·NSC,X)
(4)
根据Arrhenius公式,表面反应速率常数可通过频率因子A和活化能E这2个动力学参数来计算。然而,体系中反应规模庞大,逐一确定各反应的动力学参数难度大。对于相同类型的反应,其速率常数可通过线性自由能关联式(Linear free energy relationship, LFER)计算[10-11],如式(5)所示,其中,RI称为活性指数(Reactivity index),与参与反应各组分的电化学和热力学性质相关。
lnki=a+b·RIi
(5)
基于过渡态理论,频率因子A与反应熵变有关,而同类反应的熵变基本相同,可认为同类反应的频率因子相同[12-13],对比Arrhenius公式和式(5),同类反应的活化能与RI存在线性关系,并可以式(6)表达[13]。式(6)中,E0为各类反应的基准活化能,kJ/mol;k0为与之相对应的各类反应的基准速率常数,s-1;α为关联参数。笔者采用此方法,按反应类型管理动力学参数,这样各反应类型只需针对催化剂确定A、E0和α3个参数,缩小了动力学参数规模。
Ei=E0+α·RIi
(6)
4 半再生重整动力学模型校正及预测效果
4.1模型校正
基于上述反应网络和动力学参数获取方法,形成了分子水平催化重整反应动力学模型。模型中的相关参数需针对所用的催化剂进行校正,包括吸附平衡关联参数以及各反应类型的频率因子A、基准活化能E0和α。选取反应条件差异较大的8组实验数据(两种原料各4组)用于参数校正,参数初值及区间来源于笔者对催化重整及类似体系的研究积累。将模型计算的反应器出口产物分布与实测值比较,根据偏差情况对相关参数进行优化调整。在校正过程中主要做了以下简化处理。
(1)相比于实验测得的单体烃,模型中增加了约100个分子组分,这些组分可能存在于色谱“混合峰”中,依据沸点建立其与混合峰的映射关系,以设定其在原料和产物中的含量。
(2)重点考察模型对产物中C5+液收和C6~C9芳烃含量的预测精度,在校正目标中对其设以较高权重值。
(3)为避免模型中动力学参数相差过大而增加计算难度,烯烃组分含量直接通过化学平衡计算确定。此外,因产物中烯烃总量较低,质量分数约为1.0%,不纳入校正目标。
(4)实验测得的催化剂积炭量较低,不对生焦反应动力学参数进行校正。
表3列出了校正后各主要反应类型的频率因子和基准活化能,以及反应温度为490℃时的基准速率常数。由表3可见,芳烃脱烷基反应速率明显低于加氢裂化,将其独立为1个反应类型是合理的;芳烃甲基迁移反应速率明显高于加氢裂化和氢解,考虑此类反应对模拟芳烃组分分布是必要的;氢解反应速率较慢,反应网络限制此类反应的数量是合理的。
4.2预测效果及分析
利用其余10组实验数据检验校正模型的预测效果。图2为催化重整C5+液收和产物PINA族组成的模型计算值与实测值,图3为模型对C6~C12芳烃产率的预测效果,预测偏差列于表4。
由图2可见,模型预测的C5+液收、正构烷烃和环烷烃含量较为准确,说明模型中加氢裂化、氢解等生成轻组分的反应类型设置较为合理,链烷烃异构化反应速率比较准确;然而,模型对异构烷烃含量的预测值偏低,对芳烃含量的预测值偏高,说明异构烷烃转化为芳烃的比率高于实际情况,结合图3可知,C7~C9芳烃含量偏高尤为明显。

表3 催化重整主要反应类型的动力学参数及490℃时基准速率常数

图2 催化重整C5+液收和产物PINA族组成 实验值(wexp)与预测值(wpred)的比较Fig.2 Comparison between predicted and experimental data of C5+ yield and PINA composition of catalytic reforming
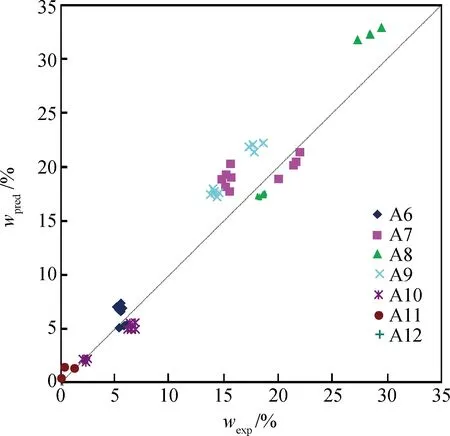
图3 催化重整C6~C12芳烃产率实验值(wexp)与 预测值(wpred)的比较Fig.3 Comparison between predicted and experimental data of C6-C12 aromatics yields in catalytic reforming 表4 催化重整C6~C9芳烃组分产率预测偏差 Table 4 Prediction error for yields of C6-C9 aromatic species in catalytic reforming

Speciesw/%MaxMinAvgSpeciesw/%MaxMinAvgBenzene1.830.171.10o-Ethyltoluene1.200.590.92Toluene4.730.552.56m-Ethyltoluene2.151.471.80Ethylbenzene1.060.180.44p-Ethyltoluene1.361.151.23o-Xylene2.020.010.911,2,3-trimethylbenzene0.760.590.67m-Xylene3.811.122.281,2,4-trimethylbenzene1.770.681.28p-Xylene1.820.000.851,3,5-trimethylbenzene0.250.000.17Propylbenzene1.290.540.97Indane0.290.120.19Isopropylbenzene1.220.731.00
从以上结果可以得出,重整体系中链烷烃脱氢环化反应速率明显低于异构化(见表3),因脱氢环化速率过高而导致链烷烃过量转化为芳烃的可能性不大,与Parera等[14]的相关结论吻合。
在分析结果中出现的链烷烃“混合峰”可能包含一定数量的多侧链异构烷烃,这些组分较难通过脱氢环化转化为芳烃,受分析水平及反应机理研究深度所限,模型尚未对此因素进行完全描述。
两种原料中C10~C12组分含量较高,质量分数约10%。对于这些未明确解析的组分,模型中以少数带较长直链的模型化合物简化表达,而其在体系中可通过加氢裂化、氢解等反应转化为C7~C9芳烃,模型化合物结构以间位为主,所以间位芳烃组分产率的预测偏差最大(见表4)。
5 结 论
(1)基于现有仪器分析水平及计算能力,建立了分子水平半再生重整反应动力学模型。包含305个C1~C12分子组分及864个化学反应,其中涵盖数十种烯烃及C6~C9全部15种芳烃组分的生成与转化。
(2)以LHHW速率方程模拟体系中的多相催化反应,采用LFER方法求取动力学参数,并针对特定催化剂对模型参数进行了校正,结果表明,芳烃脱烷基反应速率较低,而芳烃甲基迁移反应速率不容忽视,可影响芳烃产物分布。
(3)模型对贫、富芳烃2种原料在10组不同反应条件下的C5+液收、正构烷烃和环烷烃产率的预测较为准确,对C6~C9芳烃组分产率的预测偏差平均值小于3%。
(4)提高对原料和产物中单体烃,尤其是C10~C12组分的解析度,有助于更为细致地描述链烷烃与芳烃的转化过程,从而提升模型对相关组分产率的预测精度。
(5)重整体系中链烷烃异构化反应速率较快且复杂度高,深入研究双功能催化作用下的异构化反应机理,对进一步认识催化重整反应化学具有重要意义。
参考文献
[1] SMITH R B. Kinetic analysis of naphtha reforming with platinum catalyst[J].Chem Eng Prog, 1959, 55(6): 76-88.
[2] FROMENT G F. The kinetics of complex catalytic reaction[J].Chem Eng Sci, 1987, 42(5): 1073-1087.
[3] RAMAGE M P, GRAZIAZI K R. Development of Mobil’s kinetic reforming model[J].Chem Eng Sci, 1980, 35(1-2): 41-48.
[4] 周红军,石铭亮,翁惠新,等.芳烃型催化重整集总反应动力学模型[J].石油学报(石油化工),2009, 25(4): 545-550.(ZHOU Hongjun, SHI Mingliang, WENG Huixin, et al. Lumped kinetic model of aromatic type catalytic naphtha reforming[J].Acta Petrolei Sinica (Petroleum Processing Section), 2009, 25(4): 545-550.)
[5] ANTOS G J, AITANI A M. Catalytic Naphtha Reforming[M].New York, US: Marcel Dekker, 1995: 75-104.
[6] JOSHI P V, KLEIN M T, HUEBNER A L, et al. Automated kinetic modeling of catalytic reforming at the reaction pathways level[J].Rev Process Chem Eng, 1999, 2(3): 169-193.
[7] 徐承恩.催化重整工艺与工程[M].北京:中国石化出版社,2006: 168-188.
[8] KLEIN M T, HOU G, BERTOLACINI R J, et al. Molecular Modeling in Heavy Hydrocarbon Conversions[M].New York, US: CRC Press, 2006: 109-121.
[9] FROMENT G F, BISCHOFF K B. Chemical Reactor Analysis and Design[M].3rd Edition. New York, US: John Wiley & Sons, 2010: 67-86.
[10] NEUROCK M, KLEIN M T. Linear free energy relationships in kinetic analysis: Applications of quantum chemistry[J].Polyc Arom Compounds, 1993a, 3: 231-246.
[11] KORRE S. Quantitative structure/reactivity correlations as a reaction engineering tool: Applications to hydrocracking of polynuclear aromatics[D].Newark: University of Delaware, 1995.
[12] HAMMETT L P. The effect of structure upon the reactions of organic compounds. Benzene derivatives[J].J Am Chem Soc, 1937, 59(1): 96-103.
[13] DEWAR M J S. The Molecular Orbital Theory of Organic Chemistry (Advanced Chemistry)[M].New York, US: McGraw-Hill, 1969.
[14] PARERA J M, FIGOLI N S. Reactions in commercial reformer[J].Chem Ind, 1995, 61: 45-78.
收稿日期:2015-04-24
文章编号:1001-8719(2016)04-0748-06
中图分类号:TE624.4
文献标识码:A
doi:10.3969/j.issn.1001-8719.2016.04.013
Molecular Level Kinetic Model for Semi-Regenerative Reforming
ZHOU Xiang1, WANG Jieguang1, HOU Zhen2, BENNET Craing A2,MA Aizeng1, KLEIN Michael T2, GUO Jinbiao1
(1.ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China;2.EnergyInstitute,UniversityofDelaware,Newark,USA)
Abstract:Based on experiment results, a kinetic model was built in molecular level for semi-regenerative reforming involving 305 of C1-C12molecular species and 864 of chemical reactions. Formation and conversion of dozens of olefin and all C6-C9 aromatic species were considered in the model. The reaction rate laws were expressed with Langmuir-Hinshelwood-Hougen-Watson (LHHW) formalism, and kinetic parameters were obtained by Linear free energy relationship (LFER). With the analysis data of feeds and products, the model was tuned and the prediction precision of the model was tested. The results indicated that dealkylation of aromatics was slow enough to differ from hydrocracking. The rates of aromatic methyl shift reactions were not negligible and their contribution should lie in the distributions of C8+aromatics. With 2 feeds of different potential aromatic abundance processed under 10 different reaction conditions, the yields of C5+liquid products, normal paraffins and naphthenes were accurately predicted by the model, and the average yield(mass fraction) prediction errors of all C6-C9 aromatic species were under 3%.
Key words:molecular level; catalytic reforming; kinetic model; fixed bed
通讯联系人: 周祥,男,高级工程师,博士,主要从事炼油过程模型及炼油企业调度优化研究;Tel:010-82368733;E-mail:zhoux.ripp@sinopec.com
