EGFR/PI3K/AKT信号通路在肺癌中的研究进展
2016-08-10综述栾材富审校
王 娜 综述,栾材富 审校
(1.青岛大学,山东青岛 266000;2.青岛大学医学院附属烟台毓璜顶医院,山东烟台 264000)
EGFR/PI3K/AKT信号通路在肺癌中的研究进展
王娜1综述,栾材富2审校
(1.青岛大学,山东青岛 266000;2.青岛大学医学院附属烟台毓璜顶医院,山东烟台 264000)
关键词:表皮生长因子受体;磷脂酰肌醇-3激酶;AKT;肺癌;靶向治疗
肺癌是临床最常见的恶性肿瘤之一,为我国城市肿瘤相关死亡的主要病因,近年来肺癌的发病率和病死率增长迅速,严重危害人类健康和生命。在我国,每年大约有30万人被诊断患有肺癌,超过25万人因该病死亡[1]。肺癌从组织学上分为小细胞型肺癌和非小细胞型肺癌(NSCLC),其中NSCLC占85%左右,包括鳞癌、腺癌、大细胞性肺癌[2]。NSCLC的传统治疗包括放疗、化疗和手术,但是超过75%的晚期肺癌患者不适合手术[3]。近年来肿瘤的靶向治疗迅速发展,为NSCLC的治疗提供了机遇。研究发现,EGFR/PI3K/AKT信号通路的过度激活将促进肺癌细胞增殖、 侵袭及转移并抑制细胞凋亡[4]。
1EGFR/PI3K/AKT信号转导通路概述
表皮生长因子受体(EGFR)也被称为ERBB1和HER1,是一种跨膜酪氨酸激酶受体(RTK),相对分子质量为170×103,广泛分布在除造血组织细胞、体壁内胚层细胞及成熟骨骼肌细胞外的人体细胞中。EGFR是参与细胞信号通路的重要成分,由胞外区、跨膜区和胞内区三部分组成,其中胞外区主要与配体结合,而胞内区含有ATP结合区和酪氨酸激酶区。EGFR 与配体在胞外区结合后形成二聚体并使胞内区磷酸化,进而激活下游一系列信号通路。EGFR在正常的胚胎发育、组织损伤修复中发挥着重要作用,它与表皮生长因子和转化生长因子-α等共同调节人体细胞的增殖、分化、存活和转移等[5]。研究表明,EGFR 高表达可促进肿瘤细胞增殖、抑制细胞凋亡,并使血管形成加速、 细胞黏附性增强,导致肿瘤发生、侵袭和转移[6]。
磷脂酰肌醇-3激酶(PI3K)是脂类激酶家族中一员,在细胞增殖、存活等细胞功能方面具有重要的作用[7]。PI3Ks根据结构和功能分为Ⅰ、Ⅱ和Ⅲ类,Ⅰ类又分为ⅠA和ⅠB,其中ⅠA类是异质二聚体蛋白,在肿瘤形成中发挥重要作用。ⅠA类PI3Ks由p110(即PIK3CA)催化亚基和p85调节亚基构成,这两个亚基都能被表皮生长因子酪氨酸激酶(RTKs)激活。p110亚基由p110α、p110β和p110δ 3个亚型组成,分别被 PIK3CA、PIK3CB和PIK3CD基因编码;p85亚基由5个亚型p85α、p85β、 p55α、p55γ和p50α组成。
AKT是一种丝氨酸/苏氨酸激酶,也被称为蛋白激酶B(PKB),由N端PH区、中心催化区和C端调节区三部分组成。AKT能够磷酸化TSC2、FOXO蛋白、eNOS、BAD及IKKα等多种蛋白底物,是细胞增殖、存活和代谢的调节器。哺乳动物雷帕霉素靶蛋白(mTOR)是一种丝氨酸/苏氨酸激酶,属于磷脂酰肌醇激酶相关激酶(Pikk)家族,分为mTORC1和mTORC2。
EGFR与其配体结合后,引起自身磷酸化和二聚体化,激活PI3K/AKT下游信号通路,使ⅠA类PI3Ks募集到细胞膜上,使磷脂酰肌醇-4,5-二磷酸(PIP2)磷酸化为磷脂酰肌醇-3,4,5-三磷酸(PIP3),PIP3作为第二信使与AKT的PH区域结合,随后AKT的催化结构域Thr308位点被PDK1磷酸化,而C-末端疏水区域的Ser473位点则被mTORC2磷酸化,从而激活AKT。AKT活化后使结节性硬化症相关蛋白2和1(TSC2-TSC1)磷酸化而失活,从而导致Rheb GTP水平升高,激活mTORC1,mTORC1活化后磷酸化多种下游蛋白如核糖体p70S6激酶(p70S6K)、eIF4E结合蛋白(4E-BPs)等,这些效应蛋白共同促进肿瘤细胞的生长和蛋白质合成[8-10],如图1。
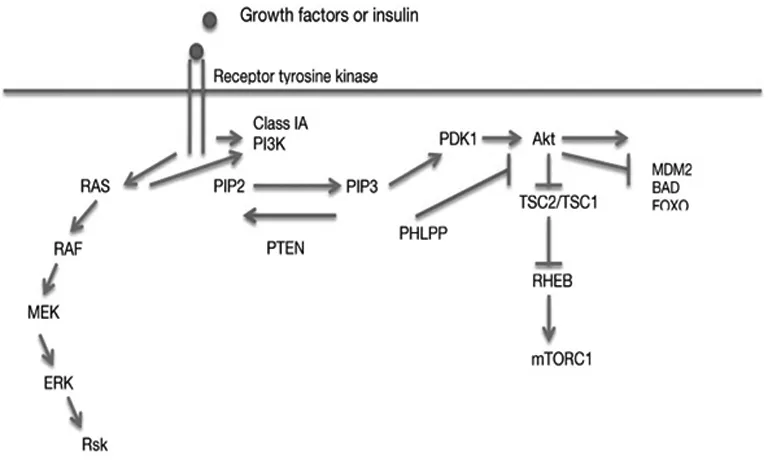
图1 EGFR/PI3K/AKT/mTOR信号通路[11]
2EGFR/PI3K/AKT信号传导通路的调控机制
EGFR/PI3K/AKT信号传导通路受多种因子的调节,如Gab1、PTEN等。Grb2相关蛋白1(Gab1)是正调控因子,在EGFR/PI3K/AKT信号通路中发挥着重要作用。当EGFR被激活后,磷酸化Gab1,招募大量的PI3K等下游信号蛋白,Gab1的PH区能特异的结合PI3K激酶产物PIP3,促使Gab1迁移到细胞膜上,增强Gab1介导的EGFR/PI3K/AKT信号通路的传导,而C端特异性酪氨酸磷酸化后可与PI3K调节亚基的p85结合,从而使PI3K大量激活,增强EGFR/PI3K/AKT信号的传递[12]。肿瘤抑制基因PTEN是EGFR/PI3K/AKT信号通路的负调控因子。PTEN具有磷酸酶活性,可以使细胞内PIP3去磷酸化形成PIP2,使其丧失第二信使作用并阻断PI3K信号通路。研究发现,PTEN表达缺失可以导致p-AKT活性增强,促进癌细胞增殖,抑制细胞凋亡[13]。此外,Zhong等[14]发现嵌合泛素连接酶通过负调控EGFR信号通路,促进EGFR泛素化和降解,阻滞下游的PI3K/AKT信号通路,抑制细胞的增殖和转移,促进细胞凋亡。Yu等[15]研究发现mTORC1磷酸化S6激酶和Grb10后能发挥负反馈效应,减少PI3K、AKT的活化,从而抑制肿瘤细胞的发展进程。
3EGFR/PI3K/AKT信号通路在肺癌中的作用机制
EGFR作为肿瘤生长的刺激物,其突变和异常过表达与肺癌的发生有关。RAS/RAF/MEK/ERKS信号通路和PI3K/AKT信号通路是EGFR突变的2个主要的信号网络系统[16-17]。其中,PI3K/AKT作为主要的信号通路可通过直接磷酸化多种转录因子促进肿瘤细胞的增殖、抑制凋亡并增强细胞的侵袭和转移能力。
3.1EGFR/PI3K/AKT信号传导通路与肺癌细胞的增殖、凋亡EGFR/PI3K/AKT信号通路是重要的抗凋亡通路,而且与新生血管的生成有关。该通路促增殖和抗凋亡机制主要包括以下3种:
3.1.1转化生长因子(TGF)途径TGF-α通过EGFR/PI3K/AKT信号通路上调Sox-2和生存素蛋白的表达,促进肿瘤细胞的凋亡[18]。Liu等[19]发现TGF-β在转录后水平上通过EGFR/PI3K/AKT抑制TGIF(TG相互作用因子)的表达,促进转录细胞凋亡。
3.1.2Notch信号途径Notch信号在调节细胞的增殖、分化、凋亡中具有重要作用。Notch家族在肿瘤细胞中主要通过EGFR/PI3K/AKT信号通路发挥作用,但是各成员扮演的角色不尽相同,Notch2上调可以抑制细胞生长和侵袭转移,介导细胞凋亡,而敲除Notch1则可以达到上述同样效果[20]。
3.1.3Bcl-2家族Bcl-2家族蛋白是EGFR信号通路与凋亡的主要联系枢纽。根据其功能和四种Bcl-2同源区域不同分为三大类,其中BH3亚家族(促凋亡蛋白)中的PUMA能够促进EGFR突变的肺癌细胞的凋亡。PI3K/AKT信号通路抑制剂能触发FOXO转录因子核转移,反式激活PUMA,促使肿瘤细胞凋亡[21]。
3.2EGFR/PI3K/AKT信号传导通路与肺癌细胞的侵袭与转移肿瘤细胞侵袭和转移到远处器官是一个复杂的过程,包括多种细胞因子、溶性生长因子、黏附受体和组织重构。EGFR/PI3K/AKT信号通路在肺癌细胞侵袭和转移中占有重要作用。上皮-间质细胞转变(EMT)是癌细胞侵袭转移的重要机制,主要表现为带有上皮标记的E-钙黏蛋白表达下降,上皮细胞间黏附性和极性降低,而代表间叶细胞性质的N-钙黏蛋白和波形蛋白表达升高。E-钙黏蛋白缺失或N-钙黏蛋白表达升高都能加速NSCLC细胞的侵袭和转移[22-23]。研究发现,EGF/EGFR信号通过激活PI3K/AKT下游通路,介导FoxO1核输出信号,激活基质蛋白酶MMP9,从而加速NSCLC的侵袭和转移[24]。Li等[25]研究发现暴露在电离辐射(IR)的肺癌患者其EGFR和整联蛋白α2β1明显升高,它们通过介导PI3K/AKT信号通路,共同促进了肿瘤细胞的侵袭和转移。有研究发现肿瘤细胞能够分泌大量的乙酰胆碱,它可以激活M3毒蕈碱受体,通过EGFR/PI3K/AKT信号通路传导促进肿瘤细胞增殖、侵袭和转移[26]。
4以EGFR/PI3K/AKT信号通路为靶点的药物在肺癌中的治疗应用
EGFR是驱动肺癌发生和发展的主要病因之一。据报道,在NSCLC中,EFGR突变频繁发生[27]。由于EGFR在肿瘤发生中有至关重要的作用,这使EFGR被作为最重要的靶点之一来治疗NSCLC。现在,已知的85%~90% EGFR突变位点位于开放阅读框的第19号外显子的E746_A750位点和第21号外显子的L858R位点[28]。携带这些突变基因的患者用EGFR-TKIs如吉非替尼、埃罗替尼或阿法替尼(二代EGFR抑制剂)进行治疗后会显著延长其无进展生存期[29]。对于携带特定EGFR突变的肺癌患者来说,吉非替尼等EGFR-TKIs无疑给他们带来了福音,遗憾的是经过一段时间治疗后,患者总是不可避免的获得耐药性[30]。研究发现,多种机制参与了NSCLC患者对EGFR-TKIs的抵抗,例如PI3K/AKT通路异常激活、MET扩增[31]、肝细胞生长因子(HGF)过表达[32]、EGFR二次突变[33]及PTEN基因缺失[34]等,其中PI3K/AKT通路异常激活是EGFR-TKIs的获得性抵抗的最重要机制。研究发现244-MPT通过降低EGFR的磷酸化,抑制PI3K/AKT下游信号通路,从而阻断了吉非替尼的抵抗[35];去甲斑蝥素(NCTD)通过抑制Met磷酸化,从而抑制PI3K/AKT下游信号通路,逆转了HGF介导的EGFR-TKIs抵抗[36]。除了单个药物的靶向治疗,多药物联合靶向治疗对耐药的肺癌患者也产生了很好的疗效。miR-34a与吉非替尼联合靶向作用于MET克服了EGFR突变型肺癌细胞中HGF介导的吉非替尼抵抗[37];Xie等[38]研究显示谷氨酰胺酶C(GAC)抑制剂968与埃罗替尼联合应用对EGFR-TKIs耐药的NSCLC患者有很好的疗效;抗EGFR突变的酪氨酸激酶抑制剂BIBW2992和抗MET扩增的酪氨酸激酶抑制剂ARQ 197联合用药,通过抑制PI3K/AKT和MEK/EPK信号通路,抑制细胞活力,控制细胞进程,从而避免了T790M-EGFR介导的埃罗替尼抵抗[39]。
5展望
肺癌作为临床常见的恶性肿瘤,在随着社会发展和环境变化的同时其发病率和病死率逐年提高,这严重威胁着人类的健康和生命。近年来,在肺癌发病机制方面的研究越来越多,但是肺癌的确切发病机制还不是十分清楚。EGFR/PI3K/AKT信号通路是肿瘤生长的重要通路,它的失调会加快细胞周期进程,促进肿瘤细胞生长并抑制细胞凋亡。阐明EGFR/PI3K/AKT信号通路在肺癌尤其是NSCLC中相互调节的机制,可能会加深对肺癌发生、发展机制的认识并且为设计NSCLC靶向药物提供依据。然而,随着基因的突变,针对EGFR突变的单靶点抗肺癌药物已逐渐出现耐药,因此,针对EGFR/PI3K/AKT信号通路的多靶点联合用药方案在肺癌治疗中开始实施并取得了良好的疗效,这为肺癌患者的治疗提供了的新发展方向。
参考文献
[1]She J,Yang P,Hong Q,et al.Lung cancer in China:challenges and interventions[J].Chest,2013,143(4):1117-1126.
[2]Sakashita S,Mai S,Ming ST.Genes and pathology of non-small cell lung carcinoma[J].Semin Oncol,2014,41(1):28-39.
[3]Wu F,Li J,Jang C,et al.The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma.[J].Int J Clin Exper Pathol,2014,7(10):6653-6661.
[4]倪琛琛,于敏,张志红.EGFR与PI3K/AKT信号通路相关蛋白在非小细胞肺癌组织中的表达及其意义[J].安徽医科大学学报,2011,46(12):1264-1266.
[5]Harari PM.Epidermal growth factor receptor inhibition strategies in on cology[J].Endocr Relat Cancer,2004,11(4):689-708.
[6]Tamás P,Solti Z,Bauer P,et al.Mechanism of epidermal growth factor regulation of Vav2,a guanine nucleotide exchange factor for Rac[J].J Biol Chem,2003,278(7):5163-5171.
[7]Vanhaesebroeck B,Stephens L,Hawkins P.PI3K signalling:the path to discovery and understanding[J].Nat Rev Mol Cell Biol,2012,13(3):195-203.
[8]Li Y,Inoki K,Guan KL.Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity[J].Mol Cell Biol,2004,24(18):7965-7975.
[9]Sarbassov DD,Guertin DA,Ali SM,et al.Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex[J].Science,2005,307(5712):1098-1101.
[10]Shaw RJ,Cantley LC.Ras,PI(3)K and mTOR signalling controls tumour cell growth[J].Nature,2006,441(792):424-430.
[11]Yip PY.Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer[J].Transl Lung Cancer Res,2015,4(2):165-176.
[12]Rodrigues GA,Falasca M,Zhang Z,et al.A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling[J].Mol Cell Biol,2000,20(4):1448-1459.
[13]Velasco A,Bussaglia E,Pallares J,et al.PI3KCA gene mutations in endometrial carcinoma:correlation with PTEN and K-RAS alterations[J].Hum Pathol,2006,37(11):1465-1472.
[14]Zhong D,Ru Y,Wang Q,et al.Chimeric ubiquitin ligases inhibit non-small cell lung cancer via negative modulation of EGFR signaling[J].Cancer Lett,2015,359(1):57-64.
[15]Yu Y,Yoon SO,Poulogiannis G,et al.Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling[J].Science,2011,332(635):1322-1326.
[16]Li H,Schmid-Bindert G,Wang D,et al.Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines[J].Adv Med Sci,2011,56(2):275-284.
[17]West L,Vidwans SJ,Campbell NP,et al.A novel classification of lung cancer into molecular subtypes[J].PLoS One,2012,7(2):e31906.
[18]Lin F,Lin P,Zhao D,et al.Sox2 targets cyclinE,p27 and survivin to regulate androgen-independent human prostate cancer cell proliferation and apoptosis[J].Cell Prolif,2012,45(3):207-216.
[19]Liu ZM,Tseng JT,Hong DY,et al.Suppression of TG-interacting factor sensitizes Arsenic trioxide-induced apoptosis in human hepatocellular carcinoma cells[J].Biochem J,2011,438(2):349-358.
[20]Xu P,Zhang A,Jiang R,et al.The different role of Notch1 and Notch2 in astrocytic gliomas[J].PLoS One,2013,8(1):e53654.
[21]Bean GR,Ganesan YT,Dong Y,et al.PUMA and BIM are required for oncogene inactivation-induced apoptosis[J].Sci Signal,2013,6(268):ra20.
[22]Mateen S,Raina K,Agarwal C,et al.Silibinin synergizes with histone deacetylase and DNA methyltransferase inhibitors in upregulating E-cadherin expression together with inhibition of migration and invasion of human non-small cell lung cancer cells[J].J Pharmacol Exp Ther,2013,345(2):206-214.
[23] Zhang X,Liu G,Kang Y,et al.N-cadherin expression is associated with acquisition of EMT phenotype and with enhanced invasion in erlotinib-resistant lung cancer cell lines[J].PLoS One,2013,8(3):e57692.
[24] Pei J,Lou Y,Zhong R,et al.MMP9 activation triggered by epidermal growth factor induced FoxO1 nuclear exclusion in non-small cell lungcancer[J].Tumour Biol,2014,35(7):6673-6678.
[25] Li X,Ishihara S,Yasuda M,et al.Lung cancer cells that survive ionizing radiation show increased integrin α2β1- and EGFR-dependent invasiveness[J].Lab Dis Paper,2013,8(8):24-47.
[26]Xu R,Shang C,Zhao J,et al.Activation of M3 muscarinic receptor by acetylcholine promotes non-small cell lung cancer cell proliferation andinvasion via EGFR/PI3K/AKT pathway[J].Tumour Biol,2015,36(6):4091-4100.
[27] Kobayashi K,Hagiwara K.Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC)[J].Targ Oncol,2013,8(1):27-33.
[28] da Cunha Santos G,Shepherd FA,Tsao MS.EGFR mutations and lung cancer[J].Annu Rev Pathol,2011,6:49-69.
[29]Yu HA,Arcila ME,Rekhtman N,et al.Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers[J].Clin Cancer Res,2013,19(8):2240-2247.
[30]Jackman D,Pao W,Riely GJ,et al.Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer[J].J Clin Oncol,2010,28(2):357-360.
[31]Engelman JA,Zejnullahu K,Mitsudomi T,et al.Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling[J].Science,2007,316(5827):1039-1043.
[32]Takeuchi S,Wang W,Li Q,et al.Dual inhibition of Met kinaseand angiogenesis to overcome HGF-induced EGFR-TKI resistancein EGFR mutant lung cancer[J].Am J Pathol,2012,181(3):1034-1043.
[33] Kobayashi S,Boggon TJ,Dayaram T,et al.EGFR mutation and resistance of non-small-cell lung cancer to gefitinib.[J].N Engl J Med,2005,352(8):786-792.
[34]Yamamoto C,Basaki Y,Kawahara A,et al.Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations[J].Cancer Res,2010,70(21):8715-8725.
[35] Zhang Y,Yao K,Shi C,et al.244-MPT overcomes gefitinib resistance in non-small cell lung cancer cells[J].Oncotarget,2015,6(42):44274-44288.
[36]Wu H,Fan F,Liu Z,et al.Norcantharidin combined with EGFR-TKIs overcomes HGF-induced resistance to EGFR-TKIs in EGFR mutant lung cancer cells via inhibition of Met/PI3k/Akt pathway[J].Cancer Chemother Pharmacol,2015,76(2):307-315.
[37]Zhou JY,Chen X,Zhao J,et al.MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting Met[J].Cancer Lett,2014,351(2):265-271.
[38] Xie C,Jin J,Bao X,et al.Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer[J].Oncotarget,2015,7(1):610-621.
[39]Qu G,Liu C,Sun B,et al.Combination of BIBW2992 and ARQ 197 is effective against erlotinib-resistant human lung cancer cells with the EGFR T790M mutation[J].Oncol Rep,2014,32(1):341-347.
DOI:10.3969/j.issn.1673-4130.2016.14.034
文献标识码:A
文章编号:1673-4130(2016)14-1984-04
(收稿日期:2016-01-11修回日期:2016-03-24)
