苯砜基羧酸酯类化合物急性毒性的预测模型
2016-07-24陈艳张昌民瞿翠玲
陈艳,张昌民,瞿翠玲
(1.徐州工程学院化学化工学院,江苏徐州221111; 2.江苏蓝丰生物化工股份有限公司,江苏新沂221400)
苯砜基羧酸酯类化合物急性毒性的预测模型
陈艳1,张昌民2,瞿翠玲1
(1.徐州工程学院化学化工学院,江苏徐州221111; 2.江苏蓝丰生物化工股份有限公司,江苏新沂221400)
用MATLAB软件编程计算了56个苯砜基羧酸酯类化合物分子电性距离矢量(mk),同时用Hyper chem 8.0程序包计算了它们的理化参数.这两类结构参数被用于建立苯砜基羧酸酯类化合物急性毒性的预测模型.通过最佳变量子集回归的方法构建多元线性回归模型:-lg EC50=4.724+30.275m7+0.061m24+6.468m85+0.880m90-0.003V-0.096(lg P)2.该模型具有良好的稳健性和较强的预测能力.以模型中的6个参数为人工神经网络(ANN)输入层,设定6∶4∶1的网络结构,用BP算法构建人工神经网络模型,其相关系数R2为0.986.结果表明,神经网络BP算法模型的预测结果优于多元线性回归模型的预测结果.
苯砜基羧酸酯;急性毒性;分子电性距离矢量;人工神经网络;定量结构-活性相关
0 引言
苯砜基羧酸酯类化合物在杀虫剂、除草剂和驱虫剂的合成以及石油、冶金工业方面有着广泛的应用[1],但此类化合物会在一定程度上污染环境,随着人们环保意识的增强,其对环境的影响也越来越受到关注.定量结构-活性相关(QSAR)研究提供了一个节省时间和资金的方法,在有机化合物对环境影响的评估方面起着重要的作用,也是对有机化学品进行危害评价的有效手段之一[2].QSAR研究的关键是分子描述符的选择,本研究以分子电性距离矢量和理化参数作为结构参数全面表征了苯砜基羧酸酯类化合物的分子结构,并以此建立了该化合物急性毒性的六元QSAR模型,其相关系数(R2)为0.896.采用误差反向传播(back-propagation,BP)算法,模型的相关性和预测能力均得到进一步的提高[3].
1 数据与研究方法
1.1 数据的来源
56种苯砜基羧酸酯类化合物的母体结构见图1,其分子结构及急性毒性数据(-lg EC50)见表1,数据来自文献[2].
1.2 结构参数的计算方法
1.2.1 分子电性距离矢量的计算方法
分子电性距离矢量是刘树深等[4-6]克服多种著名拓扑指数的局限提出的较为全面地反映分子的几何、拓扑以及电性特征的结构参数,该理论把分子中的非氢原子分成13种原子类型(Ki).
其中:Zi、Ji分别为原子的价电子层数和非氢原子i在分子中所连接的其它非氢原子的数目,例如—O—中氧原子的类型KO=4×(6-4)+2=10,羰基O中氧原子的类型KO=4×(6-4)+1=9.
按照文献[4-6]的计算方法,利用MATLAB软件编程[7-8]计算56个苯砜基羧酸酯的分子距离矢量mk,去掉无统计意义的自变量,得到44个拓扑指数作为结构参数.
1.2.2 理化参数的计算方法
采用Hyper-chem8.0程序包计算了各化合物的理化参数:V(体积)、SAA(近似表面积)、SAG(网格表面积)、lg P(辛醇水分配系数)、(lg P)2,得到5个理化参数作为结构参数.
1.2.3 多元线性回归分析和神经网络分析
将每种化合物的上述结构参数作为自变量集,苯砜基羧酸酯的急性毒性(-lg EC50)作为因变量,应用最佳子集回归的方法进行变量的压缩和筛选,选择最佳变量组合,构建相应的QSAR模型.采用逐一剔除法(leave-one-out,LOO)计算交叉验证系数(R2
CV)对模型的稳定性及预测能力进行检验,一般公认R2CV≥0.5,所建模型具有良好的稳定性及较强的预测能力[9].
采用QSAR模型中的最佳变量组合为神经网络的输入层,利用人工神经网络的BP算法,建立神经网络模型,模型的相关性显著提高.
2 多元线性回归模型的构建和分析讨论
2.1 多元线性回归模型的构建
将由上述自变量集及因变量集输入MINITAB统计软件,利用其中的Best Subsets Regression方法,选择最佳变量组合,建立最佳QSAR模型为:
其中:n、R、R2、R2adj、S、F分别为样本数、相关系数、校正判定系数、估计标准误差、Fischer检验值.用式
(2)计算所得的预测值列于表1(Prep.1),平均误差为0.154.
2.2 模型的质量检验
利用MINITAB分析软件计算模型(式(2))的交叉验证系数(R2CV)为0.844.符合R2CV≥0.5的标准,同时该模型的R2CV比R2(0.896)略小,交叉验证标准偏差SCV(0.193)比S(0.157)略大,而且所建模型的R2adj与R2CV相差为0.039,远小于0.3.这些均说明该模型具有良好的稳定性和预测能力,且没有过拟合、不存在不相关的其他变量或数据中存在离域点[10].
另外,根据一般的统计标准,一个具有良好预测能力的QSAR模型,其R2≥0.80[11].模型(式(2))的R2=0.896,反映了影响苯砜基羧酸酯化合物急性毒性的89.6%,仅有10.4%的随机因素未被揭示,因此,具有良好预测能力.
2.3 模型(式(2))的物理意义
进入模型的电性距离矢量有m7、m24、m85、m90,它们依次代表第一类碳原子(C-)与第七类氮原子(>N-)、第二类碳原子(-C-)与第十二类硫原子(>S<)、第十类氧原子(-O-)与第十三类卤原子(-X)、第十二类硫原子(>S<)与第十三类卤原子(-X)之间的相互作用.其中-CH3、-CH2-为非极性基团,-NO2、-SO2-、-O-、-X为极性基团,它们之间的相互作用分别反映了分子的疏水能力和亲水能力.模型中还有理化参数V和(lg P)2,其中(lg P)2进入模型进一步验证了疏水性对急性毒性的影响.另外,分子的体积也是影响因素之一,分子的体积越大,空间位阻效应越大,化合物越不容易进入生物的体内,则其毒性越小.
3 人工神经网络模型
人工神经网络[12-13]作为一种新的数学模型,由于具有出色的非线性函数逼近能力,可实现高符合度的网络输入符与输出目标之间的非线性映射关系[14].其中广泛使用的是Rumelhart等在1986年提出的前馈型神经计算模型和用于调节该模型神经元联结强度的误差往回传播学习算法,即著名的BP网络[15-16].本研究采用Matlab提供的神经网络工具箱中的BP算法进行建模,其中输入层单元选用对苯砜基羧酸酯急性毒性起主要作用的6个结构参数,即多元线性回归模型中的6个自变量m7、m24、m85、m90、V和(lg P)2.为了避免过训练和过拟合,根据许碌[17]和Andrea[18]的建议规则寻找最佳隐蔽层的单元数(H),即:
其中:N、M分别是样本数和网络总权重.M被定义为:
式中:I、H、Q分别为输入层、隐蔽层和输出层的单元数.由于I=6,Q=1及N=56,可得3.056<H≤4.875.所以本研究采用6∶4∶1的网络结构建立模型.
在BP算法中,为了进一步避免过拟合和过训练,将样本分为3个集:训练集、验证集和测试集,各集化合物个数依次为33个、11个、12个.由此建立的模型训练集、验证集和测试集的相关系数分别为0.991 0、0.996 4、0.997 0,总体的相关系数R为0.993 0(R2=0.986),彼此均比较接近,说明模型具有很高的稳健性.该模型给出的预测值(见表2,列于Pre.2)与实验值非常接近,平均误差为0.04,优于多元线性回归模型给出的预测值(见图2).该模型的权重和偏置列于表2.
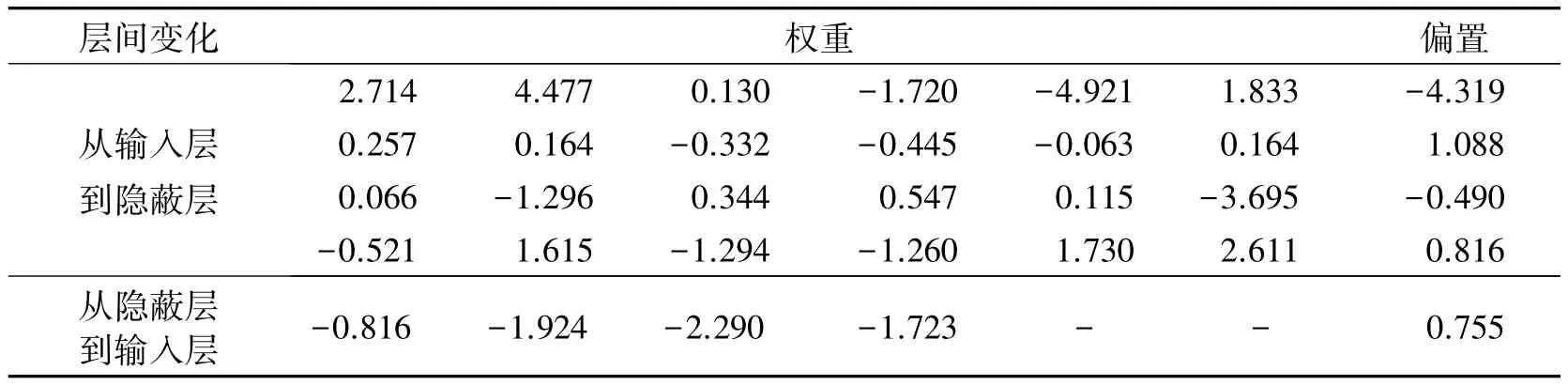
表2BP-ANN模型的权重和偏置Tab.2Weights and biases of BP-ANN model
4 结论
1)电性距离矢量和理化参数全面揭示了影响苯砜基羧酸酯急性毒性的本质因素,由进入模型的结构参数可推测苯砜基羧酸酯的急性毒性主要和疏水性及分子体积有关.
2)BP人工神经网络模型和多元线性回归模型相比具有更好的相关性、稳定性和预测能力.急性毒性和进入模型的6个结构参数呈现良好的非线性关系.利用BP-ANN预测模型预测苯砜基羧酸酯的急性毒性是可行的.本研究既可以有效地预测苯砜基羧酸酯的急性毒性,又对探讨化合物急性毒性的机理提供一定的理论依据.
[1]杭烨超,李方实.苯砜基羧酸酯类化合物急性毒性的QSAR研究[J].南京工业大学学报,2006,28(4):104-107.
[2]李吉来,杭烨超,耿彩云,等.苯砜基羧酸酯类急性毒性的QSAR研究[J].高等学校化学学报,2007,28(1):117-120.
[3]陈艳,岳玮,王彬.含氮杂环化合物理化性质和生物活性的QSPR/QSAR分析[J].武汉大学学报(理学版),2014,50(1):52-56.
[4]刘树深,刘堰,李志良,等.一个新的分子电性距离矢量(MEDV)[J].化学学报,2000,58(11):1 353-1 357.
[5]LIU S S,YIN C S,LI Z L,et al.QSAR study of steroid benchmark and dipeptides based on MEDV-13[J].Journal of Chemical Information and Computer Sciences,2001,41(2):321-329.
[6]LIU S S,LIU H L,YIN C S,et al.VSMP:a novel variable selection and modeling method based on the prediction[J].Journal of Chemical Information and Computer Sciences,2003,43(3):964-969.
[7]胡黔楠,梁逸曾,王亚丽,等.直观队列命名法的基本原理及其在矩阵与拓扑指数计算中的应用[J].计算机与应用化学,2003,20(4):386-390.
[8]张婷,梁逸曾,赵晨曦,等.基于分子结构预测气相色谱程序升温保留指数[J].分析化学,2006,34(11):1 607-1 610.
[9]DOUGLAS M H,SUBHASH C B,DENISE M.Assessing model fit by cross-validation[J].Journal of Chemical Information and Computer Sciences,2003,43(2):579-586.
[10]冯长君.手性有机酸保留指数的手性指数及原子类型电拓扑指数模型[J].物理化学学报,2010,26(1):193-198.
[11]冯子雅,冯长君.卤代苯对呆头鱼、发光菌急性毒性的构效关系研究[J].广州化工,2013,41(6):16-18.
[12]WANG B C,QIAN J Z,FAN Y,et al.The QSAR study of flavonoid-metal complexes scavenging OH free radical[J].Journal of Molecular Structure,2014,1 075(5):204-212.
[13]ZHENG F,ZHAN M,HUANG X Q,et al.Modeling in vitro inhibition of butyrylcholinesterase using molecular docking,multilinear regression and artificial neural network approaches[J].Bioorganic&Medicinal Chemistry,2014,22(1):538-549.
[14]堵锡华,史小琴,冯长君,等.基于野韭菜挥发性成分的色谱保留指数神经网络预测[J].山东大学学报(理学版),2014,49(1):468-473.
[15]冯长君,沐来龙,杨伟华,等.用拓扑指数和神经网络研究有机污染物的生物富集因子[J].化学学报,2008,66(19): 2 093-2 098.
[16]陈艳,堵锡华,吴雅萍.用拓扑指数和神经网络研究拟除虫菊酯类农药的急性毒性[J].化学通报,2014,77(4):341-345.
[17]许禄,邵学广.化学计量学方法[M].2版.北京:科学出版社,2004:441.
[18]ANDREA T A,KALAYEH H.Applications of neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors[J].J Med Chem,1991,34(9),2 824-2 836.
(责任编辑:林晓)
Prediction model of the acute toxicity of phenylsulfonyl carboxylate compound
CHEN Yan1,ZHANG Changmin2,QU Cuiling1
(1.School of Chemistry and Chemical Engineering,Xuzhou Institute of Technology,Xuzhou,Jiangsu 221111,China; 2.Jiangsu Lanfeng Bio-chem Company,Xinyi,Jiangsu 221400,China)
The molecular electrongativity distance vector(mk)and physicochemical parameters of 56 phenylsulfonyl carboxylates were calculated by the software of MATLAB and Hyper chem8.0 for establishing the prediction model of the acute toxicity(-lg EC50)of these compounds.The multiple liner regression(MLR)model was constructed by leaps-and-bounds regression:-lg EC50=4.724+30.275m7+ 0.061m24+6.468m85+0.880m90-0.003V-0.096(lg P)2.The model is highly reliable and has good predictive ability.The six structural parameters were used as the input neurons of artificial neural network,and a 6∶4∶1 network architecture was employed.A satisfied model was constructed with the back-propagation algorithm,the correlation coefficient(R2)was 0.986.It can be concluded that the prediction results of BP-ANN model are better than MLR-QSAR model.
phenylsulfonyl carboxylates;acute toxicity;molecular electrongativity distance vector; artificial neural network;QSAR
O641
A
10.7631/issn.1000-2243.2016.06.0891
1000-2243(2016)06-0891-05
2015-02-08
陈艳(1968-),教授,主要从事有机化学及物质构效学方面研究,chenyan681110@126.com
国家自然科学基金资助项目(21272095)
