基于质子传导机理的四氮唑分子间氢键研究
2016-07-22李惠萍于秀丽高鹏杰毛桢东
李惠萍, 于秀丽, 高鹏杰, 毛桢东
(郑州大学 化工与能源学院,河南 郑州450001)
基于质子传导机理的四氮唑分子间氢键研究
李惠萍, 于秀丽, 高鹏杰, 毛桢东
(郑州大学 化工与能源学院,河南 郑州450001)
摘要:为了探明四氮唑体系质子传导速率与分子间氢键强度的关系,采用密度泛函理论,在B3LYP/6-311++G(2d,2p)水平下对四氮唑二聚体N4CH2-N4CH3进行了几何结构优化和频率分析,重点分析了能量相对较低的8种二聚体的结构、能量、自然键轨道(NBO)和电荷转移量,并采用QST2方法在B3LYP/6-31+G(d)水平下研究了8种二聚体间质子传递的动态过程,发现了6种过滤态,计算了其传递能垒.结果表明,四氮唑与质子化的四氮唑阳离子通过N…H—N氢键形成分子间相互作用较强的二聚体N4CH2-N4CH3,N…H—N氢键表现出明显的红移特征;N…H—N氢键相互作用稳定化能主要是N原子孤对电子轨道与N—H键反键轨道之间的相互作用;N…H—N氢键强度是影响质子传递能垒大小的主要因素,即直接影响着四氮唑体系中质子的传导速率.
关键词:四氮唑;N…H—N氢键;密度泛函理论;质子传递
0引言
分子间相互作用对物质的性质有很大影响,在物理、化学、生物等领域发挥着至关重要的作用,对其本质的研究具有非常重要的意义[1-2].氢键是分子间相互作用的主要形式之一,是一个质子给体与一个受体之间相互作用的关系,具有较小的键能以及灵活的空间条件,可以随着分子运动不断地形成与断裂,这已成为近年来的研究热点[3].质子传递发生后,导致给体和受体的电荷和构型发生变化.如果质子传递沿氢键进行,或与相邻氢键发生耦合,则会引起体系极性的改变,产生电荷的定向传导和分子结构重排.
目前,质子交换膜燃料电池作为第三代燃料电池是世界各国研究的重点,用于质子交换膜的氮杂环类化合物具有良好的电化学稳定性和质子传导能力,在汽车和便携式电子器件方面具有很好的应用前景.有研究表明[4-6],该类质子膜在150~200 ℃及低湿甚至无水条件下,仍能表现出较高的电导率和稳定性,其质子以分子间氢键为桥梁进行传递,传导机理属于结构扩散的Grotthuss机理[7].其中,四氮唑体系因其独特的结构特点而含有丰富的氢键网络,为质子传导提供了有利条件,可表现出更强的质子传导能力.笔者选取四氮唑分子与质子化的四氮唑阳离子形成的N4CH2-N4CH3二聚物体系为研究对象,采用计算化学方法研究其氢键的结构特征和本质,为该类体系的质子传导机理研究提供理论依据.
1计算方法
密度泛函理论是一种适用于研究四氮唑体系的省时高效计算方法,可以用于氢键键能的精确计算.在B3LYP/6-311++G(2d,2p)水平下对N4CH2-N4CH3二聚体进行几何结构优化和频率分析,可以得到分子和二聚物的全优化构型.笔者分析优化得到了30种稳定的全优化构型二聚体,并重点研究了其中8种能量相对较低的主要构型如图1所示,其中a、c、d为四氮唑阳离子.首先,计算了二聚体分子间的相互作用能,并进行了基组重叠误差(BSSE)校正和零点振动能(ZPE)校正[8-9].基组重叠误差校正采用了Boys和Bernardi提出的均衡校正方法;其次,对8种二聚体进行了自然键轨道(NBO)分析计算,获得了相互作用稳定化能和分子间净电荷转移量;最后,研究了8种二聚体间质子传递的动态过程,得到了6种反应过渡态,并计算获得了反应活化能.文中所有计算均由Gaussian09程序[10]完成,二聚体相互作用能采用下述式(1)~(4)计算.
ΔE=EAB-EA-EB.
(1)
ΔEZPE=EZPEAB-EZPEA-EZPEB.
(2)
ΔECP=ΔE+EBSSE.
(3)
ΔECP+ZPE=ΔE+ΔEZPE+EBSSE.
(4)
式中:ΔE为二聚体的相互作用能;EA为四氮唑分子的单点能;EB为四氮唑阳离子的单点能;EAB为二聚体的单点能;ΔEZPE为零点振动能校正量;EZPEAB、EZPEA、EZPEB分别为四氮唑二聚体、分子、阳离子的零点振动能;ΔECP为基组重叠误差校正后的相互作用能;EBSSE为基组误差校正量;ΔECP+ZPE为二聚体经基组重叠误差与零点振动能两项校正后的相互作用能.
2结果与讨论
2.1几何结构与氢键相互作用能量
B3LYP/6-311++G(2d,2p)水平下8种四氮唑二聚体N4CH2-N4CH3全优化几何构型的相应部分键长、分子间相互作用能ΔE、经BSSE和ZPE校正后的分子间相互作用能ΔECP和ΔECP+ZPE见表1.
图1中,前6种二聚体均为平面构型(A-a-34,B-d-1,B-c-4,A-d-4,A-c-4和B-d-4),后2种为
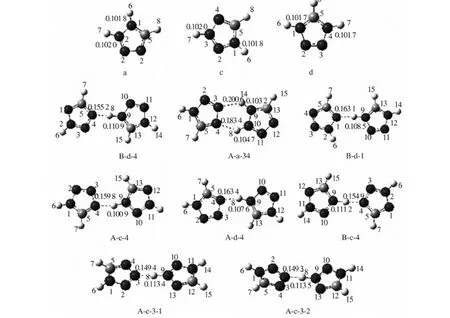
图1 四氮唑阳离子和8种二聚体的全优化几何结构(原子C:;原子N:;原子H:Fig.1 Optimized geometries of tetrazolium and eight dimers(atom C: ; H: )表1 8种N4CH2-N4CH3二聚体稳定构型的几何结构参数和相互作用能量Tab.1 Geometry parameters and interaction energy of eight N4CH2-N4CH3 stable dimers

二聚体R(N—H)/nmR’(N—H)/nmR(N…H—N)/nmΔR(N—H)/nmΔE/(kJ·mol-1)ΔECP/(kJ·mol-1)ΔECP+ZPE/(kJ·mol-1)A-a-340.10320.10180.28010.0014-141.58-139.45-134.580.10470.10200.27340.0027-141.58-139.45-134.58B-d-40.11090.10170.26610.0092-104.84-102.63-102.43B-d-10.10850.10170.27120.0068-61.63-59.67-59.54A-c-40.10890.10200.26640.0069-117.46-115.40-113.89A-d-40.10760.10170.26790.0059-115.87-113.85-111.36B-c-40.11120.10170.26600.0095-105.93-103.77-103.90A-c-3-10.11340.10200.26270.0114-120.18-117.77-118.85A-c-3-20.11350.10200.26270.0115-120.18-117.76-118.90
交叉构型(A-c-3-1和A-c-3-2),二面角约为86°.优化所得分子的几何结构参数与文献[11]所列实验数据吻合较好,键长误差大小范围为-0.01~0.03 nm;所有二聚体中的氢键均为N…H—N, 而非N…H—C.
由表1可知,与四氮唑阳离子(a、c、d)相应的N—H键长R’(N—H)相比,二聚体中的N—H键长R(N—H)均有所增大,增幅为0.001 4~0.011 5 nm,为红移特征[12].其中,二聚体A-c-3-2的N—H键长变化最大,ΔR(N—H)为0.011 5 nm;二聚体A-a-34的ΔR(N—H)最小,为0.001 4 nm.经基组重叠误差(BSSE)和零点振动能(ZPE)校正后,8种二聚体分子间相互作用能大小在-59.54-134.58 kJ·mol-1,属于强相互作用.其大小顺序为:A-a-34>A-c-3-2>A-c-3-1> A-c-4>A-d-4>B-c-4>B-d-4>B-d-1,该顺序与ΔR(N—H)和R(N…H—N)大小顺序并不一致.其中,A-a-34的ΔECP+ZPE最大,但其ΔR(N—H)最小,两个氢键键长R(N…H—N)最长,原因是A-a-34同时存在两个氢键,使其相互作用能相对较大;A-c-4和A-d-4的ΔR(N—H)明显小于B-c-4,但前两者的相互作用能却大于B-c-4,其原因可能是前者C(13)—H(15)键与相邻分子N(3)原子之间存在相互作用.
2.2自然键轨道分析
为探讨四氮唑分子间氢键相互作用的本质,还对N4CH2-N4CH3体系在B3LYP/6-311++G(2d,2p)水平下进行了自然键轨道分析(NBO),表2列出了8种N4CH2-N4CH3二聚体的自然键轨道分析的部分结果及其分子间的净电荷转移量ΔQ.对于Nx…H—N氢键体系,其相互作用的本质是Nx的孤对电子轨道和N—H的反键轨道之间的相互作用[13].稳定化能Es的大小反映了电子供体(Donor)轨道i与电子受体(Acceptor)轨道j之间相互作用的强弱.稳定化能Es越大,表示两轨道之间的相互作用越强,轨道i向轨道j提供电子的倾向越大.

表2 8种N4CH2-N4CH3二聚体的NBO分析结果及净电荷转移量
由表2可知,N4CH2-N4CH3二聚体中分子间的相互作用主要存在于四氮唑分子中N原子的孤对电子轨道n(N)和四氮唑阳离子中N—H反键轨道σ*(N—H)之间.A-c-3-2与A-c-3-1构型存在微小差别,其n(N)与σ*(N—H)间的相互作用稳定化能也非常接近,分别为320.43 kJ·mol-1、319.38 kJ·mol-1,在8种构型中最大,即其氢键作用最强.A-a-34构型两氢键的稳定化能最小,分别为42.04 kJ·mol-1和85.83 kJ·mol-1,即氢键作用最弱.8种构型的分子间氢键相互作用稳定化能的大小顺序为:A-c-3-2>A-c-3-1>B-c-4>B-d-4>A-c-4>B-d-1> A-d-4>A-a-34.这与表1中N—H键长伸长量ΔR(N—H)的大小顺序一致,符合传统的X…H—Y氢键红移特征,相互作用稳定化能越大,H—Y键伸长更明显.由电荷转移量计算结果可知,二聚体中的电子由电子供体(四氮唑分子)向电子受体(质子化的四氮唑离子)传递,其净电荷转移量ΔQ见表2.其大小顺序与稳定化能的大小顺序一致,即电荷转移量越大,相互作用稳定化能越大,氢键强度越大.
2.3 四氮唑体系中质子的传导
氮杂环类化合物作为质子载体接枝疏水性主链共聚成膜,质子载体被固定,因而质子传递可排除运载型机理,只能是结构扩散.杂环化合物分子间氢键网络重组从而引起质子在分子间传递,影响该类质子膜电导率的因素有分子间氢键的强度、聚合物主链的活动性及杂环体的含量等[14-15].
为了探明四氮唑体系质子传导率与氢键强度的关系,在B3LYP/6-31+G(d)水平下对8种二聚体间的质子传递过程进行了分析研究,反应活化能大小的主要影响因素为分子间氢键强度和两分子间转动的相对大小[16],各活化能Ea大小见表3.

表3 8种N4CH2-N4CH3二聚体在质子
A-c-3-1和A-c-3-2发生的是质子自传递反应,即反应前后二聚体构型相同.由表3可知,两者活化能相同且最小,仅为3.53 kJ·mol-1,其原因是A-c-3-1和A-c-3-2的N…H—N氢键强度最大,且为直线型,质子传递过程中两分子之间没有相对转动,反应所需的活化能较低.B-c-4和A-d-4的质子传递也属于自传递反应,活化能分别为4.45 kJ·mol-1和9.02 kJ·mol-1,其中B-c-4氢键强度稍弱于A-c-3-1和A-c-3-2,且质子传递过程中分子间发生了较小的相对转动,但B-c-4的活化能并没有明显增大,而A-d-4较大的分子间相对转动和较小的氢键强度使其反应活化能明显增大.A-a-34与B-d-1之间可以相互转化,两者氢键强度较小,反应中A-a-34两杂环间存在较大的相对转动,且需要打破其中一个氢键,因而其活化能最大.B-d-4和A-c-4之间也可以相互转化,B-d-4同样因相对较大的分子间氢键强度和较小的分子间相对转动量而具有更小的活化能3.96 kJ·mol-1,而A-c-4的活化能则为5.16 kJ·mol-1,较前者稍大.由以上分析可知,氢键作用较强的二聚体构型在质子传递反应中,分子间的相对转动量也往往较小,从而使反应所需要的活化能较低,这表明二聚体的氢键强度与其质子传递的难易程度直接相关,分子间氢键的强度是四氮唑体系质子传导快慢的主要影响因素,即强度越大,越有利于质子的传递.
3结论
(1)N4CH2-N4CH3二聚体中存在强相互作用的分子间N…H—N氢键,经基组重叠误差校正和零点能校正后的相互作用能大小在-59.54~-134.58 kJ·mol-1.
(2)分子间N…H—N氢键表现出明显的红移特征,N—H键伸长量ΔR(N—H)、轨道间相互作用的稳定化能、分子间净电荷转移量均随氢键强度的增大而增大.
(3)氢键强度是四氮唑体系质子传导快慢的主要影响因素,即强度越大,质子传递速率越快.
参考文献:
[1]吴阳,冯璐,张向东.C6H5-H…X分子间的理论计算[J]. 物理化学学报,2008,24(4):653-658.
[2]徐丽娜,肖鹤鸣,方国勇,等.NTO二聚体分子间相互作用的理论研究[J]. 化学学报,2005,63(12): 1062-1068.
[3]王庆文.有机化学中的氢键问题[M]. 天津:天津大学出版社,1993:200-201.
[4]SONG M K, LI Huiping, LIU Meilin, et al. Tetrazole based, anhydrous proton exchange membranes for fuel cells[J]. Advanced materials, 2014, 26(8), 1277-1282.
[5]BOROGLU M S, CELIK S U. Synthesis and proton conductivity studies of 5-aminotetrazole doped sulfonated polymer electrolyte membranes [J]. Polymer composites, 2011, 32(10): 1625-1632.
[6]SINIRLIOGLU D, CELIK S U, MUFTUOGLU A E. Novel composite polymer electrolyte membranes based on poly(vinyl phosphonic acid) and poly (5-(methacrylamido)tetrazole)[J]. Polymer engineering and science, 2015, 55(2):260-290.
[7]ZHOU Zhen, LIU Rui, et al. Intra- and intermolecular proton transfer in 1H(2H)-1,2,3-triazole based systems[J]. Journal of physical chemistry A, 2006, 110(7): 2322-2324.
[8]李权.三氮杂苯-水氢键复合物的结构与性质[J]. 中国科学:B辑化学,2005,35(5): 18-21.
[9]BOYS S F, BERNARDI F. The calculation of small molecular interactions by the differences of separate total energies some procedures with reduced errors [J]. Molecular physics 1970, 19(4): 553-556.
[10]FRISCH M J, TRUCKS G W, SCHLEGELH B, et al. Gaussian 09[CP].Revision A01. Wallingford CT:Gaussian Inc, 2009.
[11]ZHU Weihua, XIAO Heming. First-principles study of electronic structure, absorption spectra, and thermodynamic properties of crystalline 1H-tetrazole and its substituted derivatives[J]. Structural chemistry, 2010, 21(4): 847-854.
[12]陈自然,何展荣,张宇红. C6H5O─H…X分子间氢键的理论研究[J]. 四川师范大学学报(自然科学版),2013,36(3):435-439.
[13]肖祎,方意,刘玉震,等. 核酸中碱基的氢键作用机理及电子特征理论研究[J]. 化学研究,2014,25(2):187-194.
[14]TRUJILLO C, SANCHEZ S G. Computational study of proton transfer in tautomers of 3-and 5-hydroxypyrazole assisted by water [J]. Journal of chemical physics and physical chemistry, 2015, 16 (10):2140-2150.
[15]OZDEMIR N. Quantum chemical investigation of the intra- and intermolecular proton transfer reactions and hydrogen bonding interactions in 4-amino-5-(2-hydroxyphenyl)-2H-1,2,4-triazole-3(4H)-thione[J].Journal of molecular modeling,2013,19,(1):397-406.
[16]LI Ailin, YAN Tianying, SHEN Panwen. Exploring proton transfer in 1,2,3-triazole-triazolium dimer with ab initio method [J].Journal of power sources, 2011,196(3):905-910.
Study on the Intermolecular Hydrogen Bonds of Tetrazole Based on Proton Conduction Mechanism
LI Huiping, YU Xiuli, GAO Pengjie, MAO Zhendong
(School of Chemical Engineering and Energy, Zhengzhou University, Zhengzhou 450001, China)
Abstract:The geometric optimization and harmonic vibrational frequency for tetrazole dimers were carried out by DFT at B3LYP/6-311++G(2d,2p) level, in order to explore the relationship between the proton conducting and intermolecular hydrogen bond strength of tetrazole system. The structures, energy, natural bond orbitals and charge transfer of eight main N4CH2-N4CH3 dimers were investigated. And then, the QST2 method was employed in studying the dynamic process of proton transfer among the eight dimers at B3LYP/6-31+G (d) level. There were six transition states in the proton transfer processes among eight tetrazole dimers, and their energy barriers were also calculated. The results showed that the N…H—N hydrogen bonds of tetrazole dimers were the bridges which connected tetrazole and tetrazlium, and these hydrogen bonds showed strong intermolecular interactions and obvious red-shift characteristics. The interaction between the lone-electron pair orbital of atom N and the anti-bonding orbital of N—H was the main source of the stabilization energy. The strength of hydrogen bond was one of main factors affecting the proton transfer reaction’s energy barrier, which may directly affect the proton transfer rate of tetrazole system.
Key words:tetrazole; N…H—N hydrogen bond; DFT; proton transfer
收稿日期:2015-10-04;
修订日期:2015-12-13
基金项目:河南省国际合作交流计划项目(104300510009)
作者简介:李惠萍(1958—),女,河南洛宁人,郑州大学教授,博士,主要从事新能源材料的开发及应用研究,E-mail:huipingli@zzu.edu.cn.
文章编号:1671-6833(2016)03-0027-05
中图分类号:O641
文献标志码:A
doi:10.13705/j.issn.1671-6833.2016.03.006
