基于trnL-trnF序列的扁蓿豆和青藏扁蓿豆遗传多样性及其群体遗传结构分析
2016-07-16吴小培沈迎芳王海庆
吴小培,沈迎芳,王海庆
(1.中国科学院西北高原生物研究所高原生物适应与进化重点实验室,青海 西宁 810008;2.中国科学院大学,北京 100049)
基于trnL-trnF序列的扁蓿豆和青藏扁蓿豆遗传多样性及其群体遗传结构分析
吴小培1,2,沈迎芳1,2,王海庆1
(1.中国科学院西北高原生物研究所高原生物适应与进化重点实验室,青海 西宁 810008;2.中国科学院大学,北京 100049)
摘要:利用青藏高原及其毗邻地区的7个青藏扁蓿豆(Medicago archiducis-nicolai),以及来自上述地区和内蒙古的3个扁蓿豆(M. ruthenica)野生群体,根据叶绿体trnL-trnF基因间隔区序列,对其遗传多样性和群体遗传结构进行了分析。对161个个体的trnL-trnF序列的分析,共检测到11个核苷酸变异位点,定义了14种单倍型。对单倍型在不同群体中的分布分析显示,青藏扁蓿豆在青藏高原东南边缘地区可能存在避难所,同时在青藏高原边缘地区可能发生了青藏扁蓿豆向扁蓿豆群体的基因入侵。空间分子变异分析和基于K-2P遗传距离的群体聚类均支持将上述群体分为扁蓿豆和青藏扁蓿豆两组,组间的遗传分化程度很大。分子变异分析表明,群体内的遗传变异明显大于群体间的变异,但部分群体间存在较高水平的遗传分化;错配分布和中性检验表明,在采样范围内两种扁蓿豆都没有经历明显的近期种群扩张。研究认为,青藏高原复杂的地形结构、冰期时的气候波动以及扁蓿豆本身的进化历史可能是造成其现今遗传结构形成的主要原因。
关键词:扁蓿豆;叶绿体;trnL-trnF;遗传多样性;遗传结构
扁蓿豆(Medicagoruthenica)和青藏扁蓿豆(M.archiducis-nicolai)属于苜蓿属(Medicago)多年生植物,是阔荚苜蓿组(Section Platycarpae)的核心种[1]。扁蓿豆被认为是古北极第三纪植物区系的孑遗种[2],其种内遗传变异丰富,广泛分布于蒙古、朝鲜、俄罗斯(西伯利亚、远东地区)以及我国北方高纬度高寒地区[1,3-4],其生境多处于少雨、冬季少雪或无雪覆盖的极端寒冷地区和盐碱地带,对干旱、寒冷和盐碱胁迫具有极强的耐受性[5]。同时,由于扁蓿豆植株粗蛋白含量高、适口性好,是具有驯化潜力的多年生野生豆科牧草资源[6-7]。青藏扁蓿豆主要分布于青海、西藏及其周边毗邻的高海拔地区,是青藏高原天然高寒草地中唯一能够越冬生长的苜蓿属多年生野生豆科牧草[8]。遗传多样性是植物育种和遗传改良的基础[9]。有关扁蓿豆遗传多样性研究方面,已经有涉及形态学[10]、生化[11-12]和分子标记[13]等方面的较多报道,但研究对象大多数集中在来自内蒙古的扁蓿豆群体,对其它分布区,尤其是青藏高原及其毗邻地区扁蓿豆以及青藏扁蓿豆野生群体遗传多样性及遗传结构的研究几乎没有。
叶绿体基因组DNA为母系遗传,与核基因组相比具有较高的遗传稳定性[14],但是叶绿体的一些基因间隔区序列进化速度较快,常用于植物的系统发育研究和进化分析[15]。trnL-trnF基因间隔区为叶绿体tRNA基因trnL(UAA)(tRNAleu)和trnF(GAA)(tRNAphe)之间的非编码区,没有选择压力,片段长度适中,被广泛应用于植物系统学和谱系地理学研究[16]。本研究通过对青藏高原及其毗邻地区的7个青藏扁蓿豆,以及上述地区和内蒙古的3个扁蓿豆野生群体,共161个个体的trnL-trnF序列测序的基础上,对其遗传多样性及群体遗传结构进行分析,以期为今后开展苜蓿属野生近缘种种质资源利用及其在极端逆境下的适应机理研究提供参考信息。
1材料与方法
1.1植物材料
2012-2014年采集了位于青海和甘肃的7个青藏扁蓿豆和2个扁蓿豆,以及来自内蒙古的1个扁蓿豆野生群体(图1,表1),每个群体采集11~30个个体,群体内个体间间隔5 m以上,采集的单个个体用硅胶干燥备用。
1.2基因组DNA提取、PCR扩增及测序
每个群体随机取11~21个个体,利用CTAB法[17]提取总DNA,提取缓冲液中加入少量的PVP和β-巯基乙醇防止DNA沉淀褐化。
trnL-trnF基因间隔区序列扩增利用通用引物trnL(5′-CGAAATCGGTAGACGCTACG-3′)和trnF(5′-ATTTGAACTGGTGACACGAG-3′)[18]以及大连宝生物产品(Takara,大连)rTaq DNA合成酶进行,PCR反应体系为20 μL。扩增程序为:94 ℃预变性4 min;94 ℃变性45 s,53 ℃退火45 s,72 ℃延伸1 min,共32个循环;最后72 ℃延伸10 min。PCR扩增产物经1%琼脂糖凝胶电泳检测,确认目的条带有效扩增,且无非特异性条带后,利用上海生工生物有限公司(Sangon,上海)的柱式PCR产物纯化试剂盒进行纯化。纯化的PCR产物经过浓度检测达到测序要求后,送上海生工生物工程有限公司进行直接双向测序。
1.3数据分析
双向测序所得序列经过DNAMAN软件(http://www.shinegene.org.cn/q2.html)拼接,并根据Chromas峰图进行手工校正后,利用MEGA 6.0[19]软件中的ClustalW程序进行多序列比对,人工删除左右两边排列不整齐的序列。根据比对结果,用DnaSP v5[20]软件统计各个群体和总群体的单倍型数目(Nh)、多态位点数目(P)、核苷酸多样性(π)和单倍型多样性(hd)。对单倍型利用Network 4.2.1[21](http://www.fluxus-engineering.com)[21]软件以最大简约法构建中央连接网状图。
根据群体的遗传距离和相应的地理位置参数,利用SAMOVA 1.0[22]软件对群体进行分组,将单倍型组成和地理邻近种群进行聚类,并给出相应的FCT值(组间的遗传多样性)。组数目(K值)的取值范围为2~6,置换100次,通过寻找最大的FCT值,确定最佳的K值和分组方式。采用MEGA 6.0软件中的K-2P(kimura-2parameter)模型计算所有群体之间的遗传距离,1 000次重复的自展分析进行检验,对所得到的遗传距离矩阵同样在该软件中采用邻接法(Neighbor-Joining,N-J)进行聚类分析。用Arlequin 3.1[23]软件包计算群体间的遗传分化系数FST,通过分子变异分析(AMOVA)[24]估算遗传变异在群体内和群体间的分布,所有的计算都采用1 000次重复的置换检验检测显著性。假设trnL-trnF基因间隔区变异处于漂变-迁移平衡状态,利用Arlequin估算得到的FST值,通过公式[25-26]Nm=(1-FST)/2FST计算物种水平上居群间的平均基因流Nm[27]。

图1 10个采样群体的地理位置和14种叶绿体单倍型的分布
注:矩形框内不同的颜色代表不同的单倍型,群体编号同表1.
Note: Different colors are assigned for each population shown in the rectangle box.

表1 材料来源
为了检验群体间是否存在地理隔离效应,根据所在地点的经纬度分别计算两个种群间的地理距离矩阵,利用Arlequin计算出群体间Stakin’s[25-26]遗传距离矩阵。将上述两个矩阵导入IBDWS(http://ibdws.sdsu.edu/~ibdws/)网页在线软件检测遗传距离与地理距离的相关性,10 000次随机运算进行显著性检验。
利用DnaSP v5软件对所有群体进行基于无限突变模型的Tajima’s[28]、Fu[29]中性检验,Tajima’s D、Fu and Li’s D*和Fu and Li’s F*指数被用来推测可能的居群历史动态。显著的负值表示在发生近期居群扩张[27],正值表明居群大小相对稳定并处于动态平衡[30]。然后进一步通过错配分布分析群体的动态扩张情况。
2结果与分析
2.1trnL-trnF基因间隔区序列和单倍型分布分析
对来自7个青藏扁蓿豆和3个扁蓿豆群体共计161个个体(表1)的叶绿体trnL-trnF基因间隔区进行扩增和测序,得到长度为586 bp的序列,共检测到了11个多态位点,其中7个为简约信息位点(表2),没有发现插入/缺失片段。上述11个核苷酸变异位点在161个个体中共定义了14种单倍型(表2)。
对单倍型在不同群体内的分布分析发现,H2、H3、H7和H8的分布频率相对较高(表3)。在青藏扁蓿豆中,H2和H7单倍型为7个群体所共有,其中H2的频率相对较高。H3分布于除同德(TD)之外的6个青藏扁蓿豆群体和扁蓿豆卓尼(ZN)群体中。H8分布于乐都(LD)、内蒙古(NM)和卓尼(ZN)3个扁蓿豆群体中,且分布频率较高,是这3个群体的主要单倍型。甘南(GN)群体中包含了青藏扁蓿豆除H13外的所有单倍型类型,而在其它地点的青藏扁蓿豆群体中单倍型类型处于2~5种,因此,甘南可能存在青藏扁蓿豆的避难所。3个扁蓿豆群体中,除了H8之外,没有群体间共享的其它单倍型,显示上述3个群体可能从不同的路线迁徙而来,或者在迁徙过程中发生了较高程度的分化。此外,扁蓿豆卓尼(ZN)群体中出现了和青藏扁蓿豆部分群体共享的H3、H4和H6单倍型,表明可能在该区域发生了青藏扁蓿豆向扁蓿豆的基因入侵。其余单倍型以低频率方式只分布在少数或单个群体中。基于最大简约标准的单倍型网络分析结果显示,单倍型H2、H3、H7和H8居于网络图的相对中心位置,属于相对古老的单倍型,其余单倍型分别以一步或多步突变成为较年轻的单倍型分布在网络图的外部节点(图2A)。

表2 单倍型比对后的序列差异位点
注:所有单倍型序列参照单倍型H1,表头数字表示核苷酸变异位点。圆点表示该位点核苷酸与单倍型H1相同。Note: All sequences are compared to haplotype H1 and the number at the top indicates polymorphic sites. Dots represent nucleotide sites identical to H1.

表3 trnL-trnF序列单倍型在各个群体中的分布
2.2遗传多样性分析
对叶绿体trnL-trnF基因间隔序列单倍型多样性和核苷酸多样性的分析表明,青藏扁蓿豆群体表现出较高水平的单倍型多样性(hd=0.650)和相对较低的核苷酸多样性(π=0.001 83)。扁蓿豆群体具有相对低的单倍型多样性(hd=0.470)和高的核苷酸多样性(π=0.002 06)。在青藏扁蓿豆中,甘南(GN)群体具有最高的单倍型多样性和核苷酸多样性(hd=0.838,π=0.002 47),同德(TD)群体的遗传多样性最低(hd=0.228,π=0.000 58),其余群体介于二者之间(表4)。3个扁蓿豆群体中,可能由于基因入侵,造成了群体异质性增加,导致卓尼(ZN)群体具有相对较高的遗传多样性(hd=0.507,π=0.00282);乐都(LD)群体遗传多样性相对较低(hd=0.439,π=0.000 99),可能与该点地处农业区,受人类活动影响较大有关。
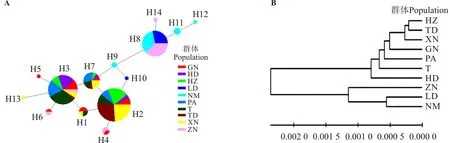
图2 单倍型网络图和群体遗传聚类图
注:A,基于最大简约标准的trnL-trnF单倍型网络图。圆圈的大小代表各单倍型的频率,矩形框内不同的颜色代表不同的群体。B,基于K-2P遗传距离的10个野生扁蓿豆群体的N-J聚类图。标尺代表遗传距离,群体编号同表1。
Note: A, Median-jointing network oftrnL-trnFhaplotypes. The size of circumference is in proportion to haplotype frequency and different colors are assigned for each population shown in the rectangle box. B, Neighbor-joining clustering of 10 populations based on K-2P genetic distance oftrnL-trnFsequences among populations.The scale is the genetic distance and population symbols are consisitent with Table 1.
2.3群体遗传聚类分析与SAMOVA分组
基于K-2P遗传距离的N-J群体聚类表明,本研究采集的10个群体在遗传距离为0.001 2处分为两个较大的分支,卓尼(ZN)、乐都(LD)和内蒙古(NM)群体聚为一支,其余群体聚为另外一支(图2B)。结合trnL-trnF序列变异和地理位置信息的SAMOVA分组结果也显示,当K=2时FCT值最大,因此,把10个采样群体划分为两个组是最佳的分组方式,来自乐都(LD)、卓尼(ZN)和内蒙古(NM)的群体归为一组,其余群体构成另一组(表5)。
2.4群体遗传结构分析
在上述分析结果基础上,对扁蓿豆和青藏扁蓿豆种间、种内群体间和群体内的分子变异分析表明,种间变异最大,占总变异成分的63.71%(FCT=0.637 12,P=0.002 93),其次为群体内变异,为总变异成分的30.15%(FST=0.698 45,P=0.000 00)(表6)。分别对扁蓿豆和青藏扁蓿豆两个种的分子变异分析发现,群体内变异成分占总变异的80%以上,群体间变异组成不到20%(表6)。由Nm=(1-FST)/2FST估算得到,青藏扁蓿豆和扁蓿豆群体间的平均基因流分别为2.047和4.065,种内群体间的基因流较大。
两两群体间的遗传分化分析结果表明(表7),群体间遗传分化系数变化较大。扁蓿豆3个群体之间,乐都(LD)和卓尼(ZN)群体与内蒙古(NM)群体之间遗传分化系数达显著水平(P<0.05),可能与其地理距离较远,产生了地理隔离有关。青藏扁蓿豆群体间遗传分化系数介于-0.0552~0.6240,部分群体间遗传分化达到了显著(P<0.05)或极显著(P<0.01)水平,可能与青藏高原及其毗邻地区的气候波动频繁以及地形复杂导致部分群体间的基因交流困难有关(表6)。
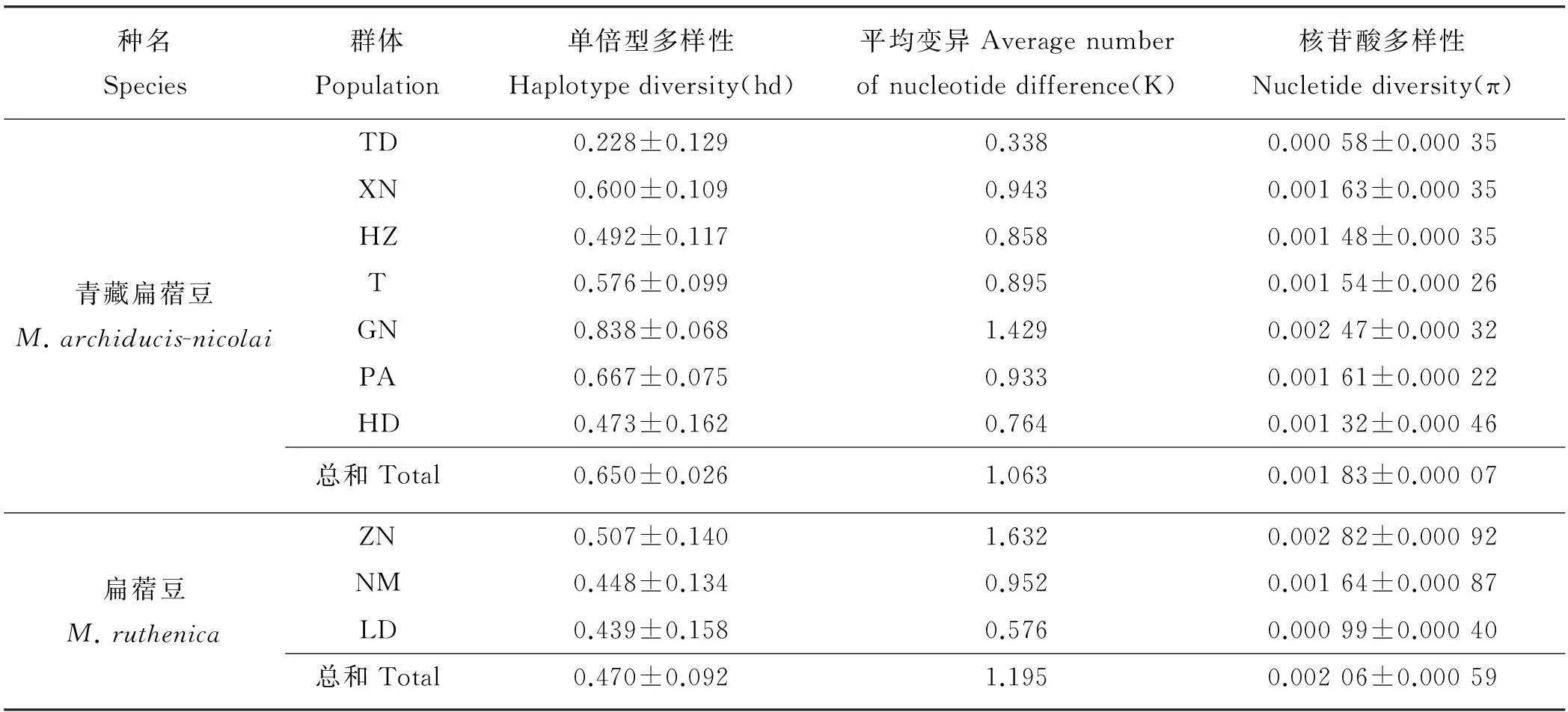
表4 青藏扁蓿豆和扁蓿豆群体群体遗传多样性指数

表5 10个群体的SAMOVA分组
2.5trnL-trnF序列中性检验及其错配分布分析
对trnL-trnF的中性检验结果表明,青藏扁蓿豆群体的Tajima’s D、Fu and Li’s F*为正值,Fu andLi’sD*为负值,但均未达到显著水平(表 8)。进一步的错配分布分析显示,采样范围内该种所有个体其观测到的错配分布曲线呈多峰曲线(图 3),表明群体处于动态平衡,没有经历明显的近期居群扩张[29],与中性检验的结果一致。扁蓿豆群体的3个中性检验值都为负值但不显著,错配分布曲线也呈现出多峰曲线,表明整体上该种群体处于动态平衡但某些群体可能经历了局部的近期动态扩张。

表6 基于trnL-trnF序列的总群体和SAMOVA分组群体的分子变异分析

表7 群体间遗传分化系数
注:**差异达极显著水平(P<0.01);*差异达显著水平(P<0.05)。
Note:** indicates significant difference at 0.01 level;* indicates significant difference at 0.05 level.

表8 基于trnL-trnF序列的青藏扁蓿豆和扁蓿豆群体的3种中性检验
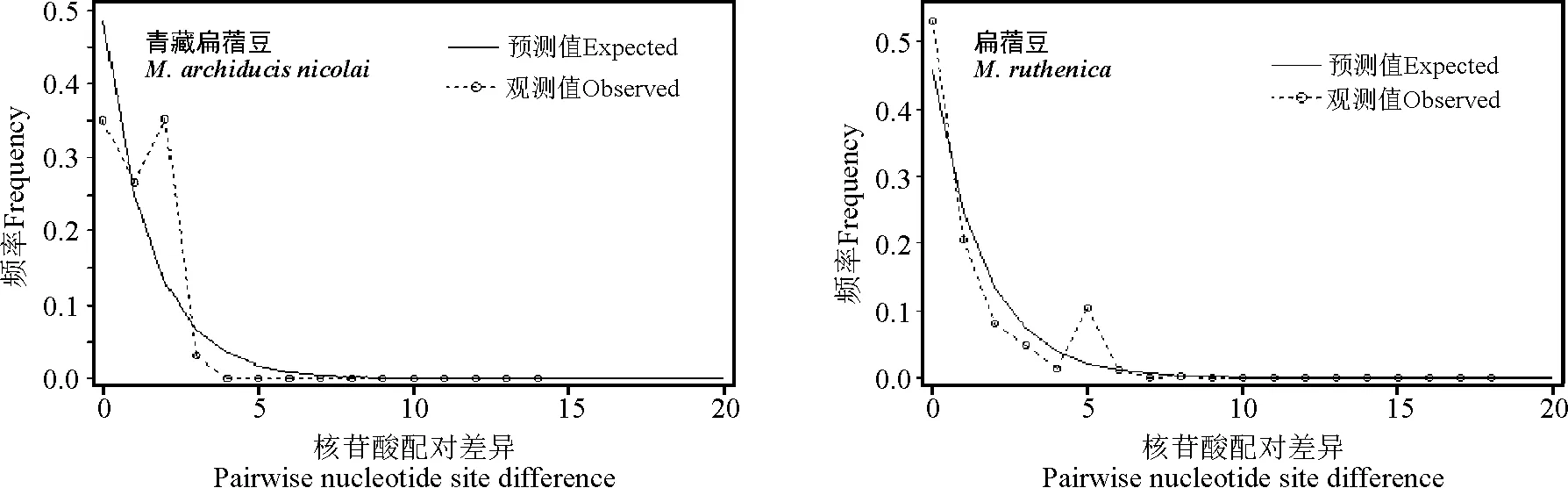
图3 基于trnL-trnF序列的两种扁蓿豆核苷酸错配分布图
3讨论
3.1野生扁蓿豆群体的遗传多样性及其形成的可能原因
此前对青藏高原地区高山绣线菊(Spiraeaalpina)和蒙古绣线菊(S.mongolica)的研究发现,物种野生群体呈现出较高的单倍型多样性和较低的核苷酸多样性,推测是由于在进化过程中发生了种群爆发而产生了众多单倍型所致[30],本研究中青藏扁蓿豆群体也出现了相似的现象。近年来,对青藏高原多个物种的研究都认为,高山植物在第四纪冰期时退缩到高原东南边缘的避难所,高原台面的居群是间冰期/冰后期由避难所居群扩散而来[31-33]。一般来说,冰期避难所居群的遗传多样性和单倍型的特有性比扩散居群要高,且冰期避难所都是比较原始的单倍型[34-35]。本研究发现,位于青藏高原东南边缘的甘南(GN)青藏扁蓿豆群体表现出较高的遗传多样性水平,推测高原东南边缘可能存在青藏扁蓿豆在第四纪冰期时的避难所,并在冰期后成为物种重新扩散的起点。同时,与甘南群体地理距离相近的卓尼(ZN)扁蓿豆群体也表现出比较高的遗传多样性,而且从中检测出了青藏扁蓿豆群体所具有的单倍型类型,表明在该区域发生了青藏扁蓿豆向扁蓿豆群体的基因入侵。但是,群体错配分布和中性检验表明,青藏扁蓿豆没有经历显著的居群扩张,遗传多样性水平也没有表现出明显的距离避难所越远遗传多样性越低的趋势。因此,我们推测,在青藏高原台面上还可能存在该物种的其它避难所。对青藏高原地区其它高山植物如银露梅(Potentillaglabra)[36-37]、西川红景天(Rhodiolaalsia)[38-39]等的研究都表明在第四纪冰期,至少是末期冰期时,这些高山植物并没有完全退却到高原东南边缘,而在高原台面上生存下来,高原台面上现有的植物分布是避难所居群在冰期结束后扩散形成。由于本研究中群体采样范围狭窄,尚无足够的证据推断哪个区域为冰期时青藏扁蓿豆在高原台面上的避难所。不过,同德(TD)群体在所有青藏扁蓿豆群体中遗传多样性水平最低(hd=0.228;π=0.000 58),这可能是冰期后居群由避难所向该区域扩散的过程中由于奠基者效应或瓶颈效应导致了遗传多样性减少。
3.2野生扁蓿豆群体的遗传结构
近年来,有关青藏高原地区植物分子系统地理学的研究发现,除了青海云杉(Piceacrassfolia)居群间的变异和居群内变异相当外,其它植物居群间的遗传变异大于居群内的遗传变异[31]。由于扁蓿豆和青藏扁蓿豆其形态上具有较高的相似性,且形态特征易受环境影响,为了减少样品采集时可能造成的分类鉴定上的偏差,本研究中首先对采集群体进行空间分子变异分组(SMOVA)和基于遗传距离的聚类分析,把扁蓿豆和青藏扁蓿豆区分开来,再进行种内的群体遗传结构分析。分子变异分析发现,两个种都表现出群体内的遗传变异大于群体间的遗传变异而且种内群体间的历史基因流较大。然而,扁蓿豆种子成熟时荚果开裂,种子依靠重力散布在母体植株周围,缺乏有效的种子传播机制[40],而且青藏高原剧烈的气候波动和复杂的地形结构也会造成群体间的基因交流障碍。在本研究中所表现出的历史基因流较大的原因可能是,扁蓿豆冰期后由避难所向外扩散过程中不同地理分布的群体汇合,发生了频繁的基因交流,而后随着群体继续向外扩散并伴随着冰期交错和气候波动,其在不同的区域又重新固定下来,但可能由于群体形成时间相对较短,产生的特有单倍型并不多,群体中主要单倍型还是祖先群体的单倍型。对青藏扁蓿豆群体遗传距离与地理距离的相关性分析表明,地理距离与群体的遗传分化没有相关性(r=-0.125 9,P=0.698 2),在很大程度上可能与所研究区域复杂的地貌结构有关,但由于采样群体数过少,依据并不充分。因此,仍需扩大其采样群体范围来进行相关性分析。
4结论
遗传多样性是种内不同种群之间或同一种群内不同个体的遗传变异的综合,是物种长期进化的产物,也是其适应、生存、进化和发展的前提[33,41]。群体的遗传结构能够反映群体在进化中所经历的突变、遗传漂变和选择效应等因素[42-43]。本研究利用叶绿体trnL-trnF基因间隔区对扁蓿豆和青藏扁蓿豆野生群体的遗传多样性进行分析,发现在青藏高原边缘可能存在青藏扁蓿豆的避难所,两个物种在青藏高原边缘存在基因入侵现象。青藏高原腹地的青藏扁蓿豆群体可能在扩散的过程中因为奠基者效应或瓶颈效应而导致了遗传多样性水平的下降。对青藏扁蓿豆和扁蓿豆群体遗传结构分析表明,由于群体间存在一定程度的历史基因流,使物种群体内的变异明显高于群体间的变异。同时,由于高原地区复杂的地形结构和剧烈的气候波动造成的基因交流障碍,导致了部分青藏扁蓿豆群体间大的遗传分化。上述研究结果为今后在大范围、大尺度上开展青藏扁蓿豆和扁蓿豆谱系地理学研究以及种质资源利用和解释其适应极端环境的响应机制提供了参考信息。
参考文献References:
[1]Small E,Jomphe M.A synopsis of the genusMedicago(Leguminosae).Canadian Journal of Botany,1989,67(11):3260-3294.
[2]Campbell T A,Bao G,Xia Z L.Agronomic evaluation ofMedicagoruthenicacollected in Inner Mongolia.Genetic Resources and Crop Evolution,1997,37:599-604.
[3]耿华珠.中国苜蓿.北京:中国农业出版社,1995:3-4.
Geng H Z.Chinese Alfalfa.Beijing:China Agriculture Press,1995:3-4.(in Chinese)
[4]郝建辉,石凤翎.不同扁蓿豆材料抗旱性比较研究.中国草地学报,2006,8(3):39-43.
Hao J H,Shi F L.Study on drought resistance ofMelilotoidesruthenicaaccessions.Chinese Journal of Grassland,2006,8(3):39-43.(in Chinese)
[5]Balabaev G A.Yellow lucernes of Siberia,Medicagoruthenica(L.) Lebd. andM.platycarpos(L.) Lebd.Bulletin of Applied Botanical Genetics and Plant Breed Services,1934,7:113-123.
[6]贾慎修.中国饲用植物志.北京:中国农业出版社,1987:337-340.
Jia S X.Flora of Chinese Forage.Beijing:China Agriculture Press,1987:337-340.(in Chinese)
[7]黄迎新,周道玮,岳秀泉,杨季云.扁蓿豆研究进展.草业科学,2007,24(12),34-39.
Huang Y X,Zhou D W,Yue X Q,Yang J Y.Research progress ofMedicagoruthenica.Pratacultural Science,2007,24(12):34-39.(in Chinese)
[8]Jin D,Ma J,Ma W,Liang C,Shi Y,He J S.Legumes in Chinese natural grasslands:Species,biomass and distribution.Rangeland Ecology and Management,2013,66(6):648-656.
[9]陈立强,师尚礼.42份紫花苜蓿种质资源遗传多样性的SSR分析.草业科学,2015,32(3):372-381.
Chen L Q,Shi S L.Genetic diversity of 42 alfalfa accessions revealed by SSR markers.Pratacultural Science,2015,32(3):372-381.(in Chinese)
[10]额尔敦嘎日迪.内蒙古中东部野生扁蓿豆形态特征多变量分析.中国草地学报,2006,28(4):87-91.
Eerdungaridi.Analysis of wildMedicagoin the middle and east Inner Mongolia mountains based on multivariate analysis of morphological characters.Chinese Journal of Grassland,2006,28(4):87-91.(in Chinese)
[11]杨青川.紫花苜蓿、扁蓿豆POD同工酶的测定.中国草地,1994(2):53-56.
Yang Q C.Studies on peroxidaseisozymes inMediacgosativacultivars andMediacgoruthenicacultivars.Grassland of China,1994(2):53-56.(in Chinese)
[12]夏兰琴.内蒙古地区扁蓿豆酯酶同工酶多样性及其地理分布的研究.中国草地,1998(2):59-63.
Xia L Q.The study on the esterase isozyme diversity ofMedicagoruthenicaand its geographical distribution in Inner Mongolia.Grassland of China,1998(2):59-63.(in Chinese)
[13]Li H Y,Li Z Y,Shi W G,Mi F G,Shi F L.Analysis of genetic diversity of Ruthenia Medic (Medicagoruthenica(L.) Trautv.) in Inner Mongolia using ISSR and SSR markers.Genetic Resources and Crop Evolution,2013,60(5):1687-1694.
[14]宫璐,李俊生,柳晓燕,赵相健,邓贞贞,赵彩云.中国沿海互花米草遗传多样性及其遗传结构.草业科学,2014,31(7):1290-1297.
Gong L,Li J S,Liu X Y,Zhao X J,Deng Z Z,Zhao C Y.Genetic diversity ofSpartinaalterniflorain coastal areas of China.Pratacultural Science,2014,31(7):1290-1297.(in Chinese)
[15]吴娟子,王强,钟小仙,陈建群.根据cpDNAtrnT-trnF序列变异分析中国互花米草种群遗传结构.草业学报,2012,21(4):134-140.
Wu J Z,Wang Q,Zhong X X,Chen J Q.Analysis of the genetic structure ofSpartinaalterniflorapopulations in China based on cpDNAtrnT-trnFsequences.Acta Prataculturae Sinica,2012,21(4):134-140.(in Chinese)
[16]狄红艳,骆凯,张吉宇,段珍,霍雅馨,王彦荣.基于ITS和trnL-trnF序列的草木樨种群遗传多样性研究.西北植物学报,2014,34(2),265-269.
Di H Y,Luo K,Zhang J Y,Duan Z,Huo Y X,Wang Y R.Genetic diversity analysis ofMelilotuspopulations based on ITS andtrnL-trnFsequences.Acta Botanica Boreali-Occidentalia Sinica,2014,34(2):265-269.(in Chinese)
[17]Doyle J J.A rapid DNA isolation procedure for small quantities of fresh leaf tissue.Phytochemical Bulletin,1987,19:11-15.
[18]Taberlet P,Gielly L,Pautou G,Bouvet J.Univeral primers for amplification of three noncoding regions of chloroplast DNA.Plant Molecular Biology,1991,17(5):1105-1109.
[19]Tamura K,Stecher G,Peterson D,Filipski A,Kumar S.MEGA6:Molecular evolutionary genetics analysis version 6.0.Molecular Biology and Evolution,2013,30:2725-2279.
[20]Librado P,Rozas J.DnaSP v5:Asoftware for comprehensive analysis of DNA polymorphism data.Bioinformatics,2009,25(11):1451-1452.
[21]Bandelt H J,Forster P,Rohl A.Median-joining networks for inferring intraspecific phylogenies.Molecular Biology and Evolution,1999,16(1):37-48.
[22]Dupanloup I,Schneider S,Excoffier L.A simulated annealing approach to define the genetic structure of populations.Molecular Ecology,2002,11(12):2571-2581.
[23]Excoffier L,Laval G,Schneider S.Arlequin(version 3.1):An integrated software package for population genetics data analysis.Evolution Bioinformatics Online,2005,1:47-50.
[24]Excoffier L,Smouse P,Quatto J.Analysis of molecular variance inferred from metric distance among DNA haplotypes:Application to human mitochondrial DNA restriction data.Genetics,1992,131(2):479-491.
[25]Slatkin M.Inbreeding coefficients and coalescence times.Genetical Research,1991,58(2):167-175.
[26]Slatkin M.A measure of population subdivision based on microsatellite allele frequencies.Genetics,1995,139(1):457-462.
[27]Nei M.Molecular Evolutionary Genetics.New York:Columbia University Press,1987:158-162.
[28]Tajima F.Statistical method for testing the neutral mutation hypothesis by DNA polymorphism.Genetics,1989,123(3):585-595.
[29]Fu Y X,Li W H.Statistical tests of neutrality of mutations.Genetics,1993,133(3):693-709.
[30]Rogers A R,Harpending H.Population growth makes waves in the distribution of pairwise genetic differences.Molecular Biology and Evolution,1992,9(3):552-569.
[31]张发起.青藏高原地区两种绣线菊的分子系统地理学和物种形成.西宁:中国科学院西北高原生物研究所博士学位论文,2012.
Zhang F Q.Molecular phylogeography and speciation of twoSpiraeaspecies(Rosaceae) on the Qing-hai-Tibetan Plateau.PhD Thesis.Xining:Northwest Insititute of Plateau Biology,2012.(in Chinese)
[32]Zhang Q,Ching T Y,George M,Liu J Q,Abbott R.Phylogeography of the Qinghai-Tibetan Plateau endemicJuniperusprzewalskii(Cupressaceae) inferred from chloroplast DNA sequence variation.Molecular Ecology,2005,14(11):3513-3524.
[33]Meng L,Yang R,Abbott R J,Miehe G,Hu T,Liu J Q.Mitochondrial and chloroplast phylogeography ofPiceacrassifoliaKom.(Pinaceae)in the Qinghai-Tibetan Plateau and adjacent highlands.Molecular Ecology,2007,16(19):4128-4137.
[34]Yang F S,Li Y F,Ding X,Wang X Q.Extensive population expansion ofPedicularislongiflora(Orobanchaceae)on the Qinghai-Tibetan Plateau and its correlation with the quaternary climate change.Molecular Ecology,2008,17(23):5135-5145.
[35]Templeton A R,Routman E,Phillips C A.Separating population structure from population history:A cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the Tiger Salaman-der,Ambystomatigrinum.Genetics,1995,140(2):767-782.
[36]Comes H P,Kadereit J W.The effect of quaternary climatic changes on plant distribution and evolution.Trends in Plant Science,1998,3(11):432-438.
[37]Wang L,Abbott R J,Zheng W,Chen P,Wang Y,Liu J Q.History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateau:Aconitumgymnandrum(Ranunculaceae).Molecular Ecology,2009,18(4):709-721.
[38]Wang L Y,Ikeda H,Liu T L,Wang Y J,Liu J Q.Repeated range expansion and glacial endurance ofPotentillaglabra(Rosaceae)in the Qinghai-Tibetan Plateau.Journal of Integrative Plant Biology,2009,51(7):698-706.
[39]Gao Q B,Zhang D J,Chen S Y,Duan Y Z,Zhang F Q,Li Y H,Chen S L.Chloroplast DNA phylogeography ofRhodiolaalsia(Crassulaceae) in the Qinghai-Tibet Plateau.Botany,2009,87(11):1077-1088.
[40]Yan J,Chu H J,Wang H C,Li J Q,Sang T.Population genetic structure of twoMedicagospecies shaped by distinct life form, mating system and seed dispersal.Annals of Botany,2009,103(6):824-834.
[41]Gao Q B,Zhang D J,Duan Y Z,Zhang F Q,Li Y H,Fu P C,Chen S L.Intraspecific divergences ofRhodiolaalsia(Crassulaceae) based on plastid DNA and internal transcribed spacer fragments.Botanical Journal of Linnean Society,2012,168(2):204-215.
[42]Soltis P S,Soltis D E.Genetic variation in endemic and widespread plant species:Examples from Saxifragageae and Polystichum(Dryopteridaceae).Aliso,1991,13(1):215-223.
[43]Slatkin M.Gene flow and the geographic structure of natural populations.Science,1987,236:787-792.
(责任编辑武艳培)
Analysis of genetic diversity and population genetic structure ofMedicagoarchiducis-nolaiandMedicagoruthenicapopulations based on cpDNAtrnL-trnFsequences
Wu Xiao-pei1,2, Shen Ying-fang1,2, Wang Hai-qing1
(1.Key Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Insititute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China;2.University of Chinese Academy of Sciences, Beijing 100049, China)
Abstract:Genetic diversity and population genetic structure of seven Medicago archiducis-nicolai and three M. ruthenica populations distributed at Qinghai-Tibet plateau(QTP)or adjacent areas and Inner Mongolia were analyzed. The results showed that, 11 polymorphic sites were detected based on the alignment of 161 trnL-trnF fragments, 14 haplotypes were identified(H1~H14). Analysis of genetic diversity and distribution of haplotypes in each population showed that the southeast edge of the Qinghai-Tibet plateau might be a refugia for M. archiducis-nicolai and gene invasion could happened across M. archiducis-nicolai to M. ruthenica in this area. Both spatial analysis of molecular variance and N-J clustering of populations based on K-2P genetic distance divided the 10 populations into two groups, that is, M. archiducis-nicolai and M. ruthenica. Analysis of molecular variation suggested that most of the genetic variation existed within populations, high population differentiation was found among some populations. Both of the two species did not experience recent population expansion based on mismatch distribution and neutrality tests. These results suggested that the genetic structure of the two species might be caused by complex topography and climate oscillations during glacial periods of the QTP. The evolutionary history of these species could also play an important role.
Key words:Medicago ruthenica; choroplast; trnL-trnF; genetic diversity; genetic structure
DOI:10.11829/j.issn.1001-0629.2015-0529
*收稿日期:2015-09-24接受日期:2016-03-11
基金项目:青海省应用基础研究计划(2014-ZJ-764)和中国科学院“西部之光”联合学者项目
通信作者:王海庆(1971-),男,青海湟中人,副研究员,博士,主要从事植物抗逆分子生物学和牧草抗逆改良研究。
中图分类号:S551+.703;Q943
文献标志码:A
文章编号:1001-0629(2016)6-1136-11*
Corresponding author:Wang Hai-qingE-mail: wanghq@mwipb.cas.cn
吴小培,沈迎芳,王海庆.基于trnL-trnF序列的扁蓿豆和青藏扁蓿豆遗传多样性及其群体遗传结构分析.草业科学,2016,33(6):1136-1146.
Wu X P,Shen Y F,Wang H Q.Analysis of genetic diversity and population genetic structure ofMedicagoarchiducis-nolaiandMedicagoruthenicapopulations based on cpDNAtrnL-trnFsequences.Pratacultural Science,2016,33(6):1136-1146.
第一作者:吴小培(1992-),女,河南方城人,在读硕士生,主要从事牧草遗传改良研究。E-mail:xiaopeiwunanyang@163.com
E-mail:wanghq@nwipb.cas.cn
