银线草HPLC指纹图谱研究
2016-07-15袁琴琴
袁琴琴
(1.菏泽学院生命科学系,山东菏泽 274000;2.陕西师范大学药用资源与天然药物化学教育部重点实验室,西北濒危药材资源开发国家工程实验室,陕西西安 710062)
银线草HPLC指纹图谱研究
袁琴琴1,2
(1.菏泽学院生命科学系,山东菏泽 274000;2.陕西师范大学药用资源与天然药物化学教育部重点实验室,西北濒危药材资源开发国家工程实验室,陕西西安 710062)
摘要[目的]建立银线草HPLC指纹图谱研究方法,为其质量控制提供依据。[方法]采用高效液相色谱法,色谱柱为Phenomenex C18(250.0 mm×4.6 mm,5.0 μm);流动相甲醇-0.1%甲酸线性梯度洗脱,流速为 0.8 mL/min;检测波长323 nm;柱温25 ℃。[结果]建立了银线草HPLC指纹图谱共有模式,并标定12个共有峰,13批样品指纹图谱相似度在0.714~0.957。[结论]不同产地的银线草化学成分存在一定差异,所建立的指纹图谱方法简单、准确可靠、重复性好,为银线草质量标准的制定提供科学依据。
关键词银线草;HPLC;指纹图谱
Key wordsChloranthusjaponicusSieb.; HPLC; Fingerprint
银线草(ChloranthusjaponicusSieb.)为金粟兰科金粟兰属多年生草本植物[1],主产于我国黑龙江、辽宁、吉林、陕西、山西和甘肃等省,以根或全草入药[2],多生于海拔500~2 300 m的山坡、山谷杂木林下阴湿处或沟边草丛中,喜富含腐殖质、湿润而排水良好的土壤[3]。由于受产地、气候和生态因子等的影响,不同产地的银线草成分有一定差异,然而仅对其主要成分的含量进行测定不能有效地控制银线草的质量。目前,指纹图谱技术已成为控制中药材质量的最重要手段之一,可以从整体上评价中药材的质量[4-5]。笔者以13个主产区的银线草为研究对象,选择迷迭香酸为参照峰,构建银线草HPLC的指纹图谱,为银线草质量标准的制定提供科学依据。
1材料与方法
1.1试验材料2011年6~8月对陕西、吉林、辽宁及黑龙江4个主产省份的银线草资源进行调查和收集,经陕西师范大学生命科学院田先华教授鉴定为金粟兰科金粟兰属植物银线草(ChloranthusjaponicusSieb.)(表1)。

表1 银线草样品的来源
1.2仪器与试剂LC2010 AHT 高效液相色谱仪(日本岛津公司),SL202N 电子秤(赛多里斯科学仪器北京有限公司),MILLI-Q 超纯水仪器(MILLIPORE公司),RE250AA 旋转蒸发仪器(上海亚荣仪器公司),KQ5200超声波清洗器(江苏昆山仪器有限公司),DFT-50手提式粉碎机(温岭市林大机械有限公司),甲醇(美国Fisher公司,色谱纯),甲酸(天津市天力化学试剂有限公司,分析纯),所用水均为超纯水。标准品:迷迭香酸(批号:07902PH,中国药品生物制品检定所),色谱柱Phenomenex C18(250.0 mm×4.6 mm,5.0 μm)(美国Phenomenex公司)。
1.3方法
1.3.1HPLC色谱条件。色谱柱采用Phenomenex C18(250.0 mm×4.6 mm,5.0 μm),流动相A:甲酸水,流动相B:甲醇;洗脱条件:0~5 min,甲醇15%~25%;5~20 min,甲醇25%~30%;20~45 min,甲醇30%~40%;45~95 min,甲醇40%~60%。流速0.8 mL/min,检测波长323 nm,柱温25 ℃,记录时间95 min;进样量10 μL。
1.3.2溶液的制备。
1.3.2.1对照品溶液的制备。精密称取迷迭香酸对照品适量,溶于甲醇溶液,浓度为0.12 mg/mL,经0.22 μm滤膜过滤后作为对照品溶液备用。
1.3.2.2供试品溶液的制备。取银线草粉末1.0 g,精密加5 mL 70%甲醇超声提取30 min,提取2次,合并提取液,摇匀,用0.22 μm微孔滤膜滤过,即得银线草样品溶液。
2结果与分析
2.1方法学考察
2.1.1精密度。精密称取S12号样品(陕西宝鸡红河谷)粉末,按照“1.3.2.2”方法进行样品提取,按照“1.3.1”的色谱条件进样5次,通过中药指纹图谱相似度计算软件计算各色谱的相似度均大于0.92,以11号峰为参照峰,12个共有峰的相对保留时间、相对峰面积的RSD均小于3%,表明该仪器的精密度良好,符合指纹图谱的要求。
2.1.2稳定性。取S13号样品(陕西宝鸡太白山)粉末,按照“1.3.2.2”方法制备供试品溶液,按照“1.3.1”的色谱条件,分别在0、4、8、12、24 h进样测定,所测得的色谱指纹图谱无明显变化,通过中药指纹图谱相似度计算软件计算各色谱的相似度均大于0.92。以11号峰为参照峰,12个共有峰的相对保留时间、相对峰面积的RSD均小于3%,表明样品溶液在24 h内稳定。
2.1.3重复性。准确称取S13号样品(陕西宝鸡太白山)粉末5份,按“1.3.2.2”方法制备样品溶液,按照“1.3.1”的色谱条件分别进样,通过中药指纹图谱相似度计算软件计算各色谱的相似度均大于0.92。以11号峰为参照峰,12个共有峰的相对保留时间、相对峰面积的RSD均小于3%,表明该方法重复性较好,符合指纹图谱的要求。
2.2银线草HPLC指纹图谱的建立与分析
2.2.1银线草指纹图谱的测定。称取13个地点的银线草粉末1 g,按“1.3.2.2”的方法制备供试品溶液,按照“1.3.1”的色谱条件依次将13批次银线草样品进行HPLC检测,将13个银线草指纹图谱导入“中药色谱指纹图谱相似度评价系统(2004A版)”进行色谱峰匹配,共有模式指纹图谱中标定12个共有峰,构成银线草指纹图谱的特征峰,并得到共有模式对照指纹图谱(图1、2) 。

图1 13批次银线草样品HPLC色谱Fig.1 The HPLC of 13 batches of Chloranthus japonicus Sieb.
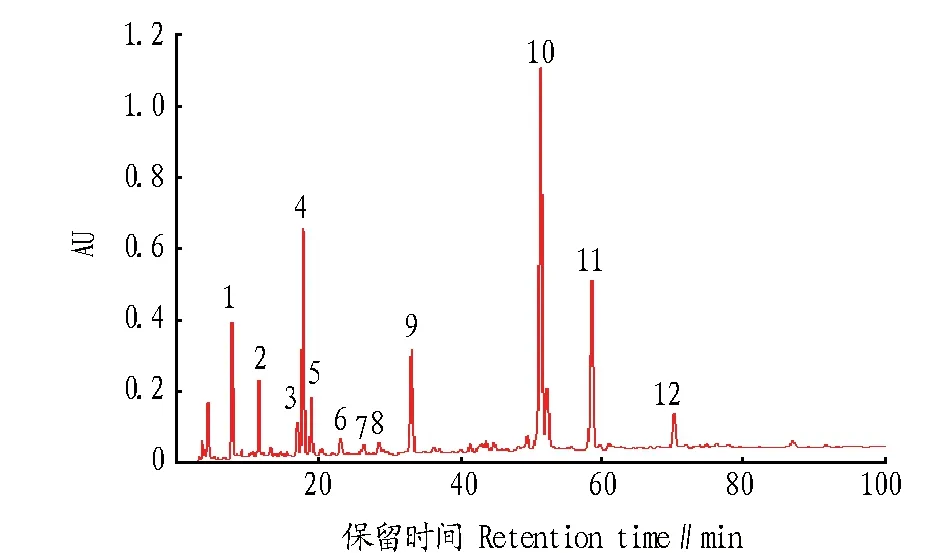
图2 共有模式对照指纹图谱Fig.2 Fingerprint of common pattern
2.2.2银线草指纹图谱的技术参数。《中国药典》(2010)规定金粟兰科草珊瑚属肿节风药材质量评价标准为异秦皮啶和迷迭香酸,且含量不低于0.02%[6-7],迷迭香酸在银线草中含量较高,远高于药典规定的0.02%,且较稳定,故选11号峰迷迭香酸为参照峰。利用国家药典委员会颁布的“中药色谱指纹图谱相似度评价系统2004A版”,依据13批次银线草供试品HPLC图谱所给出的相关技术参数,通过色谱峰的自动匹配,确定了13批次供试品的12个共有峰,即为其共有指纹峰。迷迭香酸设为参照峰S,设置参照峰的保留时间和峰面积为1.000,计算出其他共有峰的相对峰面积和相对保留时间(表2、3)。
2.2.3相似度评价。将13批次银线草的指纹图谱数据导入“中药色谱指纹图谱相似度评价系统2004A版”,以S2号批次(黑龙江省哈尔滨市帽儿山)图谱为对照图谱,使用中位数矢量法,生成了参照图谱。选择迷迭香酸这个共有峰作为校正的mark峰,生成13个产地的银线草指纹图谱。不同批次的银线草指纹图谱的相似度是以S2号银线草的图谱作为对照图谱生成的参照图谱,计算各指纹图谱间的相对保留时间和相对峰面积与参照谱图比较 。结果显示,13个产地的银线草样品指纹图谱具有较高的相似性,但也表现出一定差异,S4号吉林永吉县双河镇、S5号吉林磐石市烟筒山、S7号吉林磐石市新发北山相似度较高,在0.944以上;S6号吉林磐石市大荒山、S12陕西省宝鸡市红河谷的相似度较低;其他批次的指纹图谱相似度在0.816~0.897。
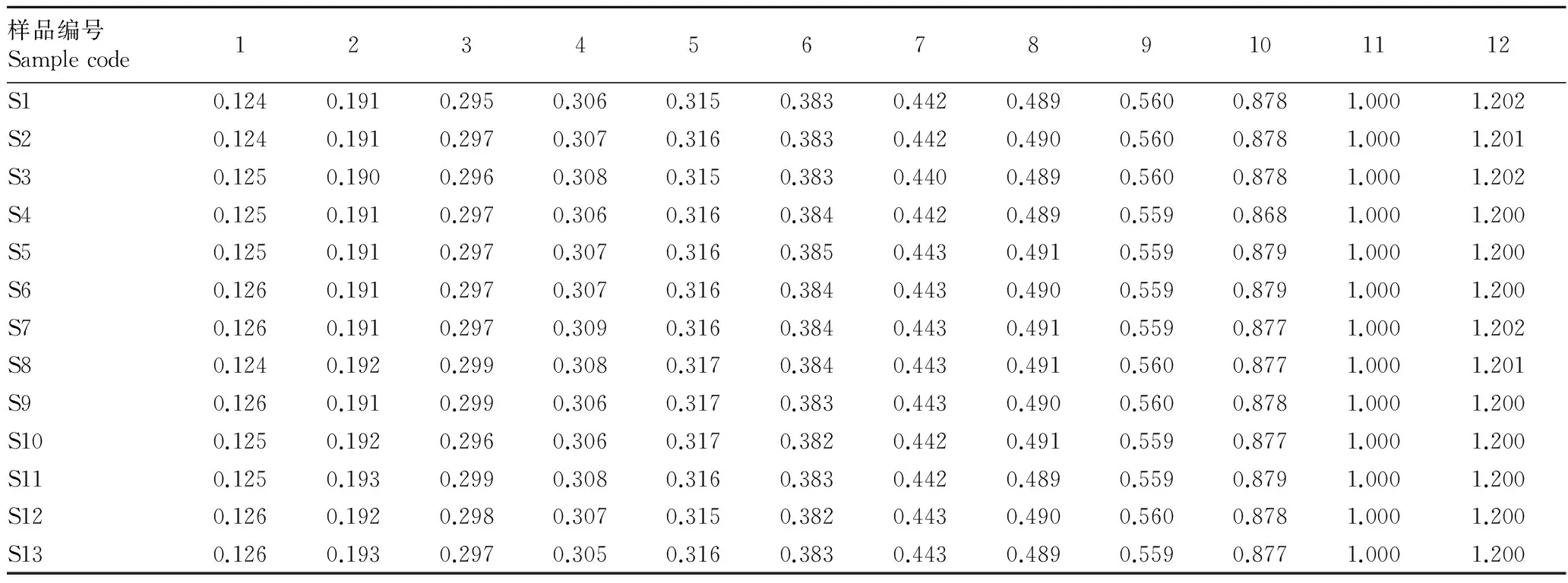
表2 银线草共有蜂的相对保留时间
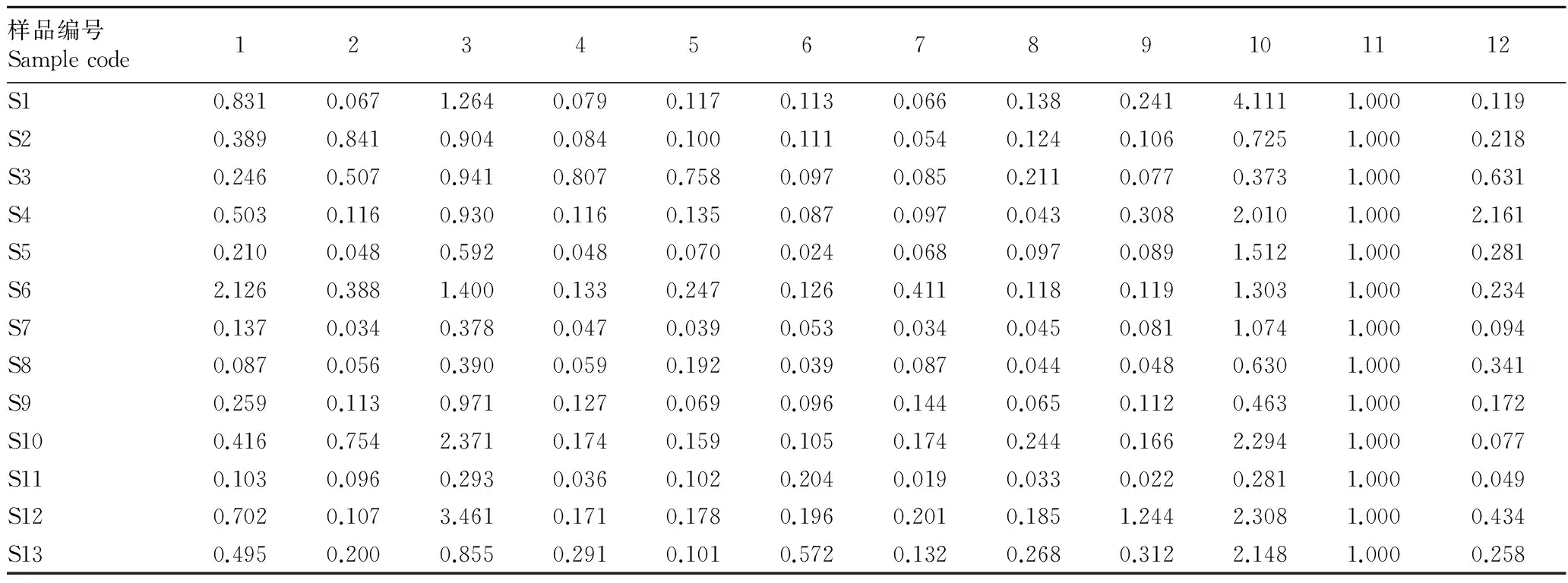
表3 银线草共有蜂的相对峰面积

表4 不同产地银线草指纹图谱相似度
3结论与讨论
该试验对银线草处理方法和HPLC色谱条件进行了优化,对不同提取溶剂及浓度进行了筛选,结果以70%甲醇提
取效果较高。在提取方法上,比较了超声与加热回流以及不同提取时间,结果表明,采用超声提取30 min提取效率较高。在流动相选择上,比较了乙腈、甲醇与水、甲酸、醋酸、磷酸溶液等组合,确定以甲醇-0.1%甲酸水溶液梯度洗脱效果最佳。在检测波长选择方面,采用DAD 检测器于 200~400 nm 下检测样品,结果显示,在 323 nm 下各峰有良好的吸收。
该研究采用HPLC方法,对陕西、黑龙江、吉林和辽宁4个主产省份银线草药材的指纹图谱进行了研究。以迷迭香酸为参照峰,13批次银线草的特征指纹图谱中有12个共有峰,尽管共有峰的保留时间相对稳定,但相对峰面积却有一定的差异,表现为各产地银线草药材的相似度降低。表明不同产地的银线草成分存在一定差异,可能由于药材受气候、海拔、产地、经纬度等生态因子的影响。
该研究以稳定性、精密度和重现性为依据,考查了银线草指纹图谱的方法学,结果表明,主要峰的相对峰面积以及相对保留时间的RSD值均低于3%,说明该方法适用于建立银线草指纹图谱,且系统稳定,符合中药指纹图谱建立的技术要求。运用相似度评价软件对银线草指纹图谱的相似度进行计算,发现13个产地的银线草指纹图谱存在一定差异,S4号吉林永吉县双河镇、S5号吉林磐石市烟筒山、S7号吉林磐石市新发北山相似度很高,在0.944以上;S6吉林磐石市大荒山、S12陕西省宝鸡市红河谷的相似度较低;其他批次的指纹图谱相似度在0.816~0.897。
该研究建立的HPLC方法能够适用于银线草的指纹图谱分析,符合中药指纹图谱建立的要求,为银线草质量标准的建立提供科学依据。
参考文献
[1] 周浙昆.金粟兰科的起源、演化及其分布[J].云南植物研究,1993,15(4):321.
[2] 陈海山,程用谦.金粟兰科的起源、分化和地理分布研究[J].热带亚热带植物学报,1994,2(4):31.
[3] 中国科学院中国植物志编辑委员会.中国植物志[M].北京:科学出版社,1982:79-80.
[4] 谢培山.中药色谱指纹图谱鉴别的概念、属性、技术与应用[J].中国中药杂志,2001,21(10):653-655.
[5] 马虹,韦永勤,林佳任.中药指纹图谱的研究及进展[J].贵阳中医学院学报,2004,26(2):58-59.
[6] 国家药典委员会.中国药典:一部 [S].北京:中国医药科技出版社,2010.
[7] 吴建章,郁建平,赵东亮.迷迭香酸的研究进展[J].天然产物研发,2005,17(3):383-388.
基金项目西安市科技创新支撑计划项目[CXY1018(3)];中央高校基本科研业务费专项资金项目(CK201002010)。
作者简介袁琴琴(1986- ),女,山东菏泽人,助教,硕士,从事植物生物技术研究。
收稿日期2016-03-26
中图分类号S 188
文献标识码A
文章编号0517-6611(2016)13-138-03
HPLC Fingerprint ofChloranthusjaponicusSieb.
YUAN Qin-qin1,2
(1. Department of Life Science, Heze University, Heze, Shandong 274000; 2. Key Laboratory of Medicinal Resource and Natural Pharmaceutical Chemistry of Ministry of Education, National Engineering Laboratory for Resource Developing of Endangered Chinese Crude Drug in Northwest of China, Shaanxi Normal University, Xi’an, Shaanxi 710062)
Key words[Objective] To establish a method of HPLC fingerprint analysis on Chloranthus japonicus Sieb., and to provide references for the quality control. [Method] High-performance liquid chromatography was adopted. The chromatographic column was Phenomenex C18(250.0 mm × 4.6 mm, 5.0 μm); mobile phase was methanol-0.1% formic acid with linear gradient elution; flow rate was 0.8 mL/min; detection wavelength was 323 nm and the column temperature was maintained at 25 ℃. [Result] HPLC fingerprint of C. japonicus was established. Twelve main marker peaks were selected. Fingerprint similarities of 13 sample batches were distributed between 0.714 and 0.957. [Conclusion] There are certain differences in the chemical components of C. japonicus in different production areas. The established fingerprint method is simple, accurate, reliable and repeatable, which provides scientific references for the quality standard establishment of C. japonicus.
