CRISPR/Cas技术可有效介导家鸡基因敲除
2016-07-14左其生王颖洁赵瑞丰程少泽汪怡临
左其生,王颖洁,赵瑞丰,程少泽,汪怡临,靳 锴,王 飞,
纪艳芹,路镇宇,张文慧,张亚妮*,李碧春*
(扬州大学动物科学与技术学院,扬州 225009)
CRISPR/Cas技术可有效介导家鸡基因敲除
左其生,王颖洁,赵瑞丰,程少泽,汪怡临,靳锴,王飞,
纪艳芹,路镇宇,张文慧,张亚妮*,李碧春*
(扬州大学动物科学与技术学院,扬州 225009)
本研究旨在家鸡上建立一种高效、稳定的基因组定点编辑技术体系,实现目的基因定点敲除,为后续家鸡基因编辑提供操作依据。基于NCBI数据库提供的CDS序列克隆C2EIP(chr2,Expression In PGC) 基因全长,并根据基因序列中APM的位置设计特异性gRNA1、gRNA2和gRNA3,并构建cas9/gRNA载体;将设计好的cas9/gRNA转染状态良好的DF-1,利用Luciferase SSA重组检测法、T7E1酶切法以及TA克隆测序法检测gRNA在DF-1细胞中基因的敲除效率。Luciferase SSA重组检测结果表明,只有cas9/gRNA3载体具有基因敲除活性,荧光活性比对照组高两倍,T7E1酶切结果显示cas9/gRNA3基因的敲除活性为27%,TA克隆测序结果表明,30个测序菌液中有8个菌液出现不同数目的碱基缺失或增加,初步估计基因敲除效率为26%。通过本研究在家鸡中初步建立了cas9介导的基因敲除技术,该技术能够稳定的在鸡的细胞DF-1上介导基因敲除。
CRISPR/Cas;基因敲除;鸡
鸡作为经典的发育生物学模型,因其鸡胚获取方便,数量大,被广泛的应用于探索基因在细胞分化过程中具体调控机制的研究[1]。家鸡的基因功能验证目前主要是通过过表达的方式,即通过干细胞介导法来产生转基因鸡或嵌合体鸡[2],但是对家鸡的基因敲除目前国内外尚无研究。
CRISPR/Cas基因敲除技术被广泛的应用于模式动物并能稳定的介导基因的敲除。P.Mali等[3]改造了cas9系统以用于人类基因组编辑,并顺利地在HEK293和K652细胞中实现基因的靶向敲除。L.Cong等[4]利用cas9系统将小鼠nero2A细胞中的Th基因以及HEK293细胞中的EMX1和PVALB基因实现定点敲除。W.Jiang等[5]与P.D.Hsu等[6]对CRISPR系统进行改造后发现,该系统可以在活细胞中启动任何基因的编辑,该系统还可以鉴定涉及特定疾病的基因,同时利用该系统鉴定了黑色素瘤细胞在抵抗癌症药物反应过程中的几个相关基因。CRISPR系统不仅可以调控基因的表达,还可以以敲入的方式校正基因的表达,Y.Wu等[7]利用CRISPR系统成功将小鼠白内障遗传病治愈,G.Schwank等[8]利用该技术校正人干细胞中一种与囊肿性纤维化相关联的基因缺陷。虽然cas9介导的基因编辑工具已经广泛的用于人[9]、小鼠[10]、斑马鱼[11-12]等物种,而且在植物上也有广泛的应用[13],但是该方法在家禽细胞中的应用尚未见报道。
鸡作为家禽生物的代表,一直被应用于生殖细胞发育分化的研究。本实验室一直致力于鸡雄性生殖细胞分化调控机制的研究,目前已完成鸡雄性生殖细胞发育分化过程中的转录组测序,并对调控这一过程的关键基因进行了筛选[14],为了进一步研究这些基因的功能,对这些基因进行基因组水平编辑已成为必要。基于此,本试验拟在鸡上建立一种以CRISPR/Cas系统介导的能够稳定敲除基因的新方法,为鸡中进行基因敲除提供操作依据,并能为构建鸡雄性生殖细胞分化的调控网络,揭示生殖细胞形成机制提供试验基础。
1 试验材料与方法
1.1试验材料
DF-1细胞由本实验室购买并保存;Luciferase-SSA载体由本试验构建并保存;VK001-08(gRNA/Cas9表达质粒,CU6启动gRNA表达,CMV启动Cas9蛋白表达)购自北京唯尚立德公司。
1.2试验方法1.2.1C2EIP基因克隆与靶位点选择通过NCBI提供的CDS序列,设计引物克隆候选基因全长。引物序列,F:5′-GAGGCTATCAAATGGCAG-3′,R:5′-ACACCCAATGAAAATAAAT-3′。根据PAM定位克隆的CDS序列,筛选并设计3个靶位点序列,分别命名为gRNA1、gRNA2和gRNA3,并将靶位点序列进行BLAST比对,确保gRNA的靶向性,不会靶向作用其他无关基因。具体信息见表1。
表1gRNA靶位点核苷酸序列
Table 1Nucleotide sequence of gRNA to target site

名称NamegRNA序列SequenceofgRNAPAMgRNA1CTTTTCTGTGCCATTCTCCAAGGgRNA2AGCACAGAGGAGTTCCTCTGAGGgRNA3TTGGATTGACATTACTCTGTAGG
1.2.2cas9/gRNA表达载体构建根据选择的gRNA分别设oligo引物,引物分别为5′AAACACCG-gRNA-1,5′CTCTAAAAC-gRNA-1-anti;5′AAACACCG-gRNA-2,5′CTCTAAAAC-gRNA-2-anti;5′AAACACCG-gRNA-3,5′CTCTAAAAC-gRNA-3-anti。步骤1:将正义链和反义链进行退火处理形成双链,形成oligo二聚体,退火体系为正、反义链各1 μL,Solution I(2×) 1 μL,H2O 3 μL。退火程序:95 ℃ 5 min,自然冷却至16 ℃,16 ℃ 10 min。步骤2:oligo二聚体插入载体,反应体系为:VK001-8载体 1 μL,oligo二聚体2 μL,H2O 7 μL。反应程序:25 ℃静置5 min。取步骤2的最终产物5~10 μL加入到刚解冻的50 μL DH5α感受态细胞中,轻弹混匀,冰浴30 min后,42 ℃热激90 s,冰上静置2 min,直接涂于氨苄抗性的平板。 12 h后挑3~5个白色菌落摇菌,提取质粒DNA进行测序。测序引物:sqprimer:TGAGCGTCGATTTTTGTGATGCTCGTCAG。
1.2.3DF-1细胞培养与转染DF-1细胞为鸡胚胎成纤维细胞的细胞系,在含有15%胎牛血清和双抗的DMEM中生长,细胞培养过程中1~2 d换液1次,当细胞的汇合度达到85%左右时,用PBS洗涤3遍,胰酶消化3 min,加入有血清的培养基终止消化,消化后的细胞收集到15 mL离心管中,1 200 r·min-1离心6 min,离心后弃上清,用新鲜培养基重悬吹打200次,接种至24孔板,接种细胞数为105个。第2天当细胞生长汇合度达60%左右进行转染试验,转染试剂为Fugene,转染条件为Fugene(V)∶质粒(μg)=3∶1,转染后48 h进行流式细胞分选获取细胞并提取基因组。
1.2.4Luciferase-SSA报告载体活性检测将终止子和靶位点插入Luciferase载体中,靶位点位于终止子前,以FUGENE为转染试剂共转染CRISPR/gRNA载体,以及新构建的Luciferase报告基因和内参donor质粒,对照组转染空载体,检测Luciferase信号(图1)。具体的工作原理:在cas9/ sgRNA的作用下,靶点位置将会产生DSB,细胞通过同源重组方式修复DNA,形成一个有活性的Luciferase,通过多功能酶标仪检测Luciferase活性。

图1 Luciferase-SSA报告载体活性检测结构示意图Fig.1 The structural representation of the detection of Luciferase-SSA activity
1.2.5T7E1酶切试验 T7 核酸内切酶 I (T7 endonuclease I,T7E1 )能够识别不完全配对DNA并对其进行切割,可用于检测CRISPR/Cas9介导的基因突变。对去转染后流式细胞分选的细胞提取基因组,以未转染的细胞为对照。根据靶位点在基因组中的位置在靶位点前后各250 bp左右设计引物,并扩增总长500 bp左右的片段,引物序列,F:5′CCTGCCCTTTACTTCGGGG3′,R:5′TGTTCCTCAAAATGCCGTGG3′,对扩增获得的片段进行98 ℃解链,自然冷却至室温,添加T7E1酶进行酶切,酶切体系为T7E1酶 1 μL,buffer 1 μL,扩增片段800 ng,H2O补足10 μL。酶切条件:37 ℃ 30 min,4 ℃保存。酶切后通过2%的琼脂糖凝胶电泳观察试验结果。计算公式:突变产物剪切条带灰度值/(突变产物剪切条带灰度值+未突变条带灰度值)。
1.2.6TA克隆测序分析CRISPR/Cas9基因敲除载体转染状态良好的DF-1,48 h后流式细胞术筛选出GFP阳性细胞,提取基因组DNA,设计引物,克隆出靶位点前后各250 bp共计500 bp左右片段,进行TA克隆,挑菌测序并与原始序列进行对比,计算CRISPR/Cas9基因敲除载体的具体效率,计算方法:序列发生突变的菌液数/全部测序菌液数×100%。
2 结 果
2.1C2EIP基因克隆与cas9/gRNA表达载体构建
以鸡原始生殖干细胞cDNA为模板,根据NCBI数据库C2EIP的CDS序列设计引物克隆该基因,序列全长为714 bp(图2A),经测序结果正确。根据测序后的序列设计3条靶位点,同时设计靶位点序列的oligo引物,通过退火杂交并与cas9/gRNA表达载体进行连接,测序结果表明gRNA1、gRNA2和gRNA3已经完整的连入载体(图2B)。
2.2Luciferase-SSA报告载体法检测cas9/gRNA载体基因敲除活性
为了检测构建的cas9/gRNA基因敲除载体是否具有敲除活性,将敲除载体与Luciferase-SSA报告载体以及donor载体共转染DF-1细胞(图3A),48 h后收集细胞检测Luciferase活性,结果显示,gRNA1和gRNA2载体Luciferase的活性与未转染组之间没有显著差异,但是gRNA3的荧光活性值与对照组相比增加2倍左右(图3B)。这说明cas9/gRNA-3载体能够有效的敲除Luciferase-SSA报告载体中对应的靶位点,Luciferase-SSA报告载体通过同源末端重组形成完整的Luciferase。

A.C2EIP克隆结果;B.gRNAs(下划线)连入cas9/gRNA载体后的测序结果A.The cloning result of C2EIP;B.The result of the gRNAs(with underline) connected into cas9/gRNA vector successfully图2 C2EIP基因克隆和cas9/gRNA载体构建Fig.2 C2EIP gene cloning and construction of cas9/gRNA vector
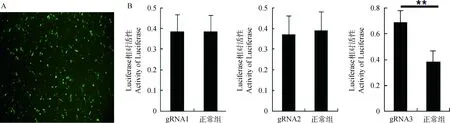
A.cas9/gRNA基因敲除载体转染DF-1细胞示意图(200×);B. Luciferase-SSA报告载体法检测结果。**.P<0.01A. The cas9/gRNA knockout vector transfected into DF-1(200×); B. The result of SSA .**.P<0.01图3 cas9/gRNA载体基因敲除活性检测Fig.3 Activity detection of cas9/gRNA vector
2.3T7E1酶切检测靶向C2EIP cas9/gRNA3的活性
将cas9/gRNA3载体通过脂质体转染DF-1,48 h后收集细胞,提取基因组,通过PCR扩增gRNA3附近424 bp片段,通过T7E1酶切方法进一步检测cas9/gRNA3载体的活性。从图4可以看出,在cas9/gRNA3试验组能清楚的检测到酶切后200 bp左右的片段,而在对照组则没有,将cas9/gRNA3试验组和对照组的样进行混合后也能观察到200 bp左右的片段。通过条带的灰度值估算活性约为27%。
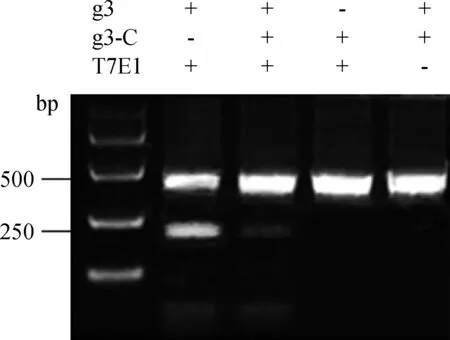
图4 T7E1酶切检测结果Fig.4 The result of T7E1 enzyme digestion
2.4TA克隆测序分析cas9/gRNA3基因敲除效率
在试验2.3中克隆纯化424 bp片段,进行TA载体连接,转化至DH5α并在氨苄培养基上涂板,14 h左右进行挑菌测序,测序结果表明,测序的30管菌液中有8管菌液发生了突变,初步估计基因敲除效率为26%左右(图5)。

图5 TA克隆测序检测cas9/gRNA3载体的基因敲除效率Fig.5 Sequencing of TA clone dected the activity of cas9/gRNA3
3 讨 论
从CRISPR/Cas发现至今,这项技术已经被广泛的用在动物的细胞水平或者个体水平的基础研究,也已经在HEK293细胞、iPS等细胞中产生了稳定的敲除细胞株,另外科研人员也利用了显微注射等方法获得小鼠、大鼠和斑马鱼等模式动物的敲除个体并顺利产生后代。M.Jinek等[15-16]以人类细胞为试验材料进行基因的靶向修饰,试验中筛选的细胞顺利产生DSB,说明cas9系统可应用于人类细胞。S.W.Cho等[17-18]以人的细胞为试验材料,通过试验证明在共转染体系中gRNA浓度的增加能够提高基因的敲除效率,最高效率达到33%。P.Mail[3]等人改造了cas9系统以用于人类基因组编辑,并顺利地在HEK293和K652细胞中实现基因的靶向敲除。L.Cong等[4]利用cas9系统在小鼠nero2A细胞中的Th基因以及HEK293细胞的EMX1基因和PVALB基因实现定点敲除。P.Mali等[3]通过试验表明,gRNA在结构上越接近crRNA和tracrRNA复合体,则在试验过程中能够获得较高的敲除效率。目前利用cas9进行基因敲除的技术已经日渐成熟,利用此技术在细胞中可实现单基因或者多基因的敲除。P.Mail等[3]利用此技术在人的细胞中实现了基因的敲除,靶向敲除效率:HEK 293为10%~25%,K562为8%~13%,iPS为2%~49%;H.Yang等[19]通过共转染的方法在小鼠的胚胎干细胞中对Tet1、Tet2、Tet3、Uty和Sry等基因实现了单基因、双基因及多基因的敲除,敲除效率达40%左右,并通过测序、RFLP以及DNA印迹等方法进行了验证。Z.Zhang等[14]也在人和小鼠的细胞中实现了多基因的同时敲除。多基因敲除试验的成功将有利于科研人员在体内研究冗余基因以及上位基因的功能。Z.Zhang等[14]通过该技术在不同细胞上研究后认为,Cas9/gRNA技术能够用于任何细胞并完成基因编辑,然而其所用的试验材料都以哺乳动物建系的细胞为试验基础,在其他物种上并未进行系统的验证,尤其是鸟类。
为了验证cas9/gRNA系统能否在家禽上对基因进行精确的基因敲除,本试验以鸡胚成纤维建系细胞DF-1为试验材料,系统的验证了cas9/gRNA系统在家禽细胞中介导基因敲除情况。试验结果表明,cas9/gRNA系统在鸡的DF-1 细胞中能够介导基因敲除,且基因的敲除效率为26%~27%,这表明该系统在家鸡细胞中能够稳定应用。虽然目前大量的研究表明,cas9/gRNA在哺乳动物、植物上介导基因的敲除效率能够达到40%~80%,但是由于本试验首次在鸡上进行,技术和细胞质量成为限制本试验基因敲除效率的重要因素。本试验的研究结果能够充分的表明cas9/gRNA系统能够在鸡上进行基因编辑,介导基因敲除,这为cas9/gRNA系统的应用前景开辟了一个全新的领域,也进一步的为基因功能验证提供了一种新的方法和思路。
[1]PAIN B,CLARK M E,SHEN M,et al.Long-terminvitroculture and characterisation of avian embryonic stem cells with multiple morphogenetic potentialities[J].Development,1996,122(8):2339-2348.
[2]LI B,SUN G,SUN H,et al.Producing transgenic chicken in swarm mediated by spermatogonial stem cellsinvivoandinvitro[J].ScienceinChina,2008,38(7):626-634.
[3]MALI P,YANG L,ESVELT K M,et al.RNA-guided human genome engineering via Cas9[J].Science,2013,339(6121):823-826.
[4]CONG L,RAN F A,COX D,et al.Multiplex genome engineering using CRISPR/Cas systems[J].Science,2013,339(6121):819-823.
[5]JIANG W,BIKARD D,COX D,et al.RNA-guided editing of bacterial genomes using CRISPR-Cas systems[J].NatBiotechnol,2013,31(3):233-239.
[6]HSU P D,LANDER E S,ZHANG F.Development and applications of CRISPR-Cas9 for genome engineering[J].Cell,2014,157(6):1262-1278.
[7]WU Y,LIANG D,WANG Y,et al.Correction of a genetic disease in mouse via use of CRISPR-Cas9[J].CellStemCell,2013,13(6):659-662.
[8]SCHWANK G,KOO B K,SASSELLI V,et al.Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients[J].CellStemCell,2013,13(6):653-658.
[9]FU Y,FODEN J,KHAYTER C,et al.High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J].NatBiotechnol,2013,31(9):822-826.
[10]LI D,QIU Z,SHAO Y,et al.Heritable gene targeting in the mouse and rat using a CRISPR-Cas system[J].NatBiotechnol,2013,31(8):681-683.
[11]HWANG W Y,FU Y,REYON D,et al.Efficient genome editing in zebrafish using a CRISPR-Cas system[J].NatBiotechnol,2013,31(3):227-229.
[12]XIAO A,WANG Z,HU Y,et al.Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish[J].NucleicAcidsRes,2013,41(14):e141.
[13]SHAN Q,WANG Y,LI J,et al.Targeted genome modification of crop plants using a CRISPR-Cas system[J].NatBiotechnol,2013,31(8):686-688.
[14]ZHANG Z,ELSAYED A K,SHI Q,et al.Crucial genes and pathways in chicken germ stem cell differentiation[J].JBiolChem,2015,290(21):13605-13621.
[15]JINEK M,CHYLINSKI K,FONFARA I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J].Science,2012,337(6096):816-821.
[16]JINEK M,JIANG F,TAYLOR D W,et al.Structures of Cas9 endonucleases reveal RNA-mediated conformational activation[J].Science,2014,343(6176):1247997.
[17]CHO S W,KIM S,KIM J M,et al.Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease[J].NatBiotechnol,2013,31(3):230-232.
[18]CHO S W,KIM S,KIM Y,et al.Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases[J].GenomeRes,2014,24(1):132-141.
[19]YANG H,WANG H,SHIVALILA C S,et al.One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering[J].Cell,2013,154(6):1370-1379.
(编辑郭云雁)
CRISPR/Cas Techniques Can Knockout the Gene of Chicken Effectively
ZUO Qi-sheng,WANG Ying-jie,ZHAO Rui-feng,CHENG Shao-ze,WANG Yi-lin,JIN Kai,WANG Fei,JI Yan-qin,LU Zhen-yu,ZHANG Wen-hui,ZHANG Ya-ni*,LI Bi-chun*
(CollegeofAnimalScienceandTechnology,YangzhouUniversity,Yangzhou225009,China)
This study aimed to establish a highly efficient and stable gene editing technique mediated by CRISPR/Cas to knock out the targeted gene,and explore its application in chicken preliminarily,and to provide the operation basis for the subsequent poultry genetic edition.We cloned the full-length ofC2EIP(chr2,Expression in PGC) gene according to sequence in NCBI Database,and designed 3 gRNAs named gRNA1,gRNA2 and gRNA3 based on the location of APM in the sequence to construct the cas9/gRNA vector.SSA activity assay,T7E1 digestion method and sequencing of TA cloning were used to detect the knock-out efficiency of the gRNA after the cas9/gRNA vector transfected into DF-1.Results of SSA activity assay showed that only cas9/gRNA3 vector could knock out the gene effectively,fluorescence activity which transfected cas9/gRNA3 was twice higher than the control group;Result of T7E1 digestion showed that activity of cas9/gRNA3 was 27%,sequencing of TA cloning results showed there were 8 mutational samples in 30 samples,and the efficiency of gene-knockout was 26%.In this study we established the technology of gene knockout mediated by CRISPR/Cas in chicken,which can be used to konck out genes in DF-1 stably.
CRISPR/Cas;gene knockout;chicken
10.11843/j.issn.0366-6964.2016.06.024
2015-10-08
国家自然科学基金(31301959;31272429;31472087);高等学校博士学科点专项科研基金资助课题(20123250120009);中国博士后基金(2012M511326;2014T70550);江苏高校优势学科建设工程资助项目;大学生创新创业训练计划项目(201411117033Z)
左其生(1992-),男,江苏盐城人,博士生,主要从事动物胚胎工程与遗传工程研究,E-mail:744366503@qq.com
李碧春,教授,博士,主要从事动物胚胎工程与遗传工程研究,E-mail:yubcli@yzu.edu.cn;张亚妮,副教授,博士,主要从事动物胚胎工程与遗传工程研究,E-mail:ynzhang@yzu.edu.cn
S831.2
A
0366-6964(2016)06-1266-06
