臭氧氧化苯并噻吩及其衍生物的反应机理的量子化学研究*
2016-07-14蒋敏结王寒露周建敏广东石油化工学院化学工程学院广东茂名525000
蒋敏结,王寒露,梁 炜,周建敏(广东石油化工学院,化学工程学院,广东 茂名 525000)
臭氧氧化苯并噻吩及其衍生物的反应机理的量子化学研究*
蒋敏结,王寒露†,梁 炜,周建敏
(广东石油化工学院,化学工程学院,广东 茂名 525000)
摘 要:采用密度泛函理论DFT/B3LYP的方法,以6-31G*以及6-311++G (d, p) 的基组对臭氧氧化苯并噻吩及其衍生物反应中的几何构型、反应路径及前线分子轨道进行了理论研究。结合实验数据,结果表明:臭氧最高分子轨道(HOMO)和苯并噻吩的最低空分子轨道(LUMO)对称性匹配而络合,经氧化分解并得到了最终稳定的氧化产物2-巯基苯甲醛。本文进一步计算了臭氧氧化二苯并噻吩和4,6-二甲基二苯并噻吩的反应机理,这两种硫化物先被氧化成亚砜,然后被进一步氧化成砜。根据所需活化能垒的大小,可得出臭氧和苯并噻吩及其衍生物的反应活性规律:苯并噻吩 > 4,6-二甲基二苯并噻吩 > 二苯并噻吩。
关键词:臭氧氧化;苯并噻吩;反应机理;氧化活性;DFT
0 引 言
氧化脱硫技术是一种十分具有前景的油品深度脱硫技术,因其在常温常压下即可反应,且不需要消耗大量的氢气而备受关注。过氧化氢是分子氧化脱硫技术中运用最广泛的氧化剂,但其单独使用过氧化氢的氧化效果较差,而在过渡金属或酸性介质脱硫体系下,过氧化氢才能表现出较为理想的氧化效果[1-2]。臭氧是一种不需添加其他催化剂的绿色的强氧化剂,氧化电位高达2.07 V,分解后的产物为水和氧气,对环境十分友好。因此臭氧无论是在石化行业还是污水治理等行业都具有十分光明的工业生产前景[3]。随着密度泛函理论的发展,量子化学研究在分子氧氧化油品中有机硫、原子掺杂以及烯烃加氢中均有应用[4-7]。但臭氧脱除油品中苯并噻吩类的有机硫化物的反应机理的量子化学研究报道较少。臭氧氧化反应迅速,反应多伴随着初级臭氧化以及次级臭氧化,很难从实验中探究其反应机理。但臭氧十分容易与含有不饱和键的烃类发生臭氧分解反应[8-15]。20世纪 50年代,CRIEGEE[16]提出如图1所示的反应机理。该机理已被实验证明。苯并噻吩的噻吩环上存在一不饱和双键,具有未成对电子。因此,可能路径如图2中的Path 1所示:IM_0 →IM_1→TS1→IM_2→TS2→IM_3→TS3→IM_4→TS4→IM_5。根据张伟[17]的实验,气相色谱-质谱仪分析结果显示苯并噻吩与臭氧的反应产物为2-巯基苯甲醛。
另外,根据前人的实验成果,分子氧氧化苯并噻吩类衍生物的氧化产物为砜[17-19]。由于苯环的共轭作用,苯并噻吩类衍生物更加容易氧化生成砜。因此推测臭氧氧化苯并噻吩的另一可能反应路线如图2中的Path 2:臭氧先氧化苯并噻吩上的硫原子生成亚砜(BTO),进一步氧化生成砜(BTO2)。本文通过量子化学计算出臭氧氧化苯并噻吩及其衍生物的反应机理,以期为臭氧氧化脱硫技术提供理论依据。
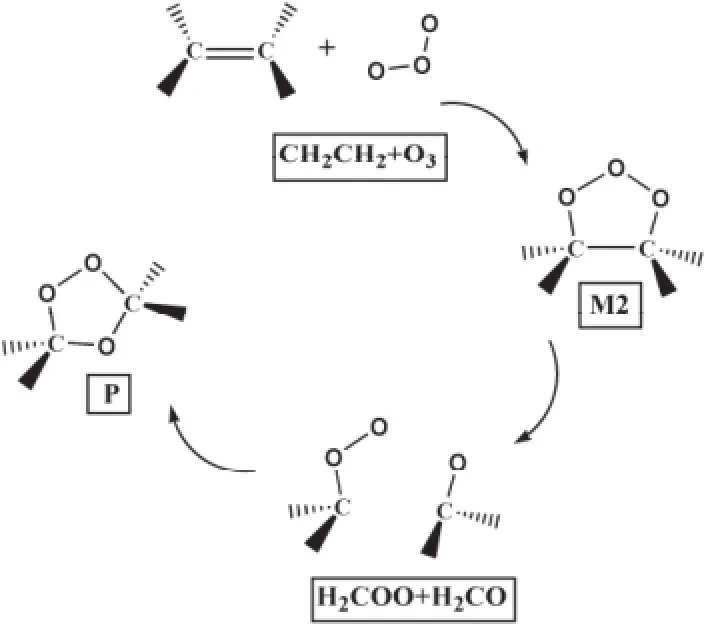
图1 Criegee机理Fig. 1 Criegee mechanism
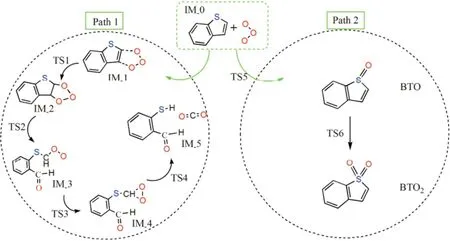
图2 臭氧和苯并噻吩反应的可能机理Fig. 2 Possible reaction mechanism of benzothiophene and ozone
2 计算方法
量子化学计算中,计算模型和基组选择会影响结果的准确性[20]。本文运用密度泛函 DFT/B3LYP方法[20-21],在6-311++G(d, p)的水平上研究了臭氧和苯并噻吩及其衍生物反应机理,优化反应物,产物以及中间体的几何构型,通过振动分析确定了优化结构为中间体(没有虚频)和过渡态(唯一虚频)。并计算出臭氧进攻苯并噻吩双键时的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)及其能隙。所有过渡态均进行了内禀反应坐标分析(IRC),以证明其过渡态的真实性。反应中各稳定点的能级用包含熵效应的吉布斯自由能(ΔG)描述,对能量的讨论均在6-311++G(d, p)基组上进行。以上计算均由Gaussian 09[22]程序完成。
3 结果与讨论
3.1 臭氧氧化苯并噻吩的反应机理
3.1.1 前线分子轨道理论
HOMO和LUMO对臭氧和苯并噻吩反应分子性质的影响非常重要。如图3所示,根据前线分子轨道理论,臭氧在进攻苯并噻吩上的双键时,首先是通过臭氧的最高占据分子轨道(π 轨道)和苯并噻吩的最低未占据分子轨道(π*轨道)对称性匹配,生成IM_1这一步计算出的放热有6.9 kcal·mol-1,对于生成IM_2的结构定位起着关键性作用。
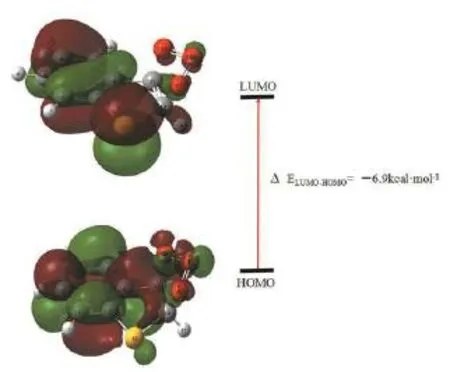
图3 臭氧和苯并噻吩的前线分子轨道Fig. 3 The frontier molecular orbital of ozone and benzothiophene
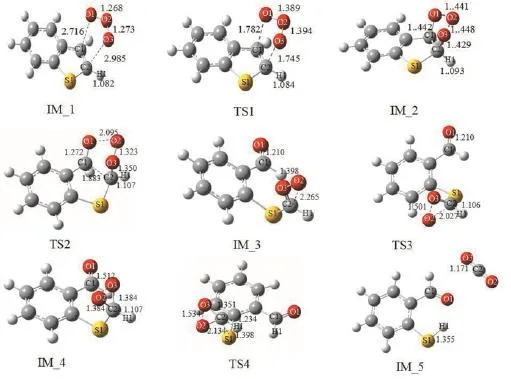
图4 臭氧和苯并噻吩反应的各个过渡态的几何构型(Path 1)Fig. 4 The geometries of the transition state of the reaction of ozone and benzothiophene ( Path 1 )
3.1.2 几何构型优化及振动分析
对各反应的反应物、中间体及产物的几何构型进行全优化。结果表明,所有驻点的力常数矩阵本征值全为正值,这表明它们在势能面上是稳定存在的点。图4列出了臭氧氧化苯并噻吩生成2-巯基苯甲醛的反应机理。反应第一步符合Criegee机理,首先 π 络合物IM_1中的O1和O3进攻苯并噻吩噻吩环双键上的C1和C2原子,经过过渡态TS1生成双五元环的中间体IM_2。通过频率分析,TS1的振动模式对应,C1=C2双键断裂,同时C1-O1和C2-O3成键,都从2.716 Å缩短为1.442 Å,O1-O2和O2-O3键长变长。O1-O2由1.268 Å 变长为1.441 Å,O2-O3 由 1.273 Å 变长为 1.448 Å。该反应所需活化能为6.6 kcal·mol-1;接着,中间体IM_2上的O1-O2键和C1-C2键断裂,C1-C2由最初的1.568 Å伸长为1.937 Å,同时O1-O2键由最初的 1.080 Å伸长为2.118 Å,导致IM_2的双五元环断裂,经过渡态 TS2分解为含有羧基基团和羰基基团的中间体IM_3。据实验结果,TS2所需活化能为13.0 kcal·mol-1。因苯并噻吩具有高的空间位阻,反应第三步并没有类似于Criegee反应机理。第三步反应进一步生成羧基基团和羰基基团π络合物。IM_3中O2进攻C2成键,由原来的2.265 Å 缩短为1.384 Å ,经过过渡态TS3生成含有三元环的中间体IM_4。TS3所需活化能为11.1 kcal·mol-1。最后,IM_4经历质子转移过程并生成最终产物2-巯基苯甲醛和CO2。通过频率分析,TS4的振动模式对应C2-S1键断裂,S1-H1成键,H1从C2原子处转移到S1原子处,该反应所需活化能为17.1 kcal·mol-1,和前文提及到张伟的实验数据吻合[17]。如图 5,从苯并噻吩和臭氧的反应机理的整体势能面来看,Path 1得到的四步活化能中,最后一步的活化能最大,为17.1 kcal·mol-1,其他三步的活化能较小,因此Path 1的控制步骤为中间体M3经过过渡态TS4生成最终产物2-巯基苯甲醛和二氧化碳的过程。最近报道的臭氧和α-蒎烯反应的能垒为22.0 kcal·mol-1[23]。另外,根据李来才的报道[24],在臭氧氧化乙烯的反应中,其控制步骤所需要的活化能为17.2 kcal·mol-1,与臭氧氧化苯并噻吩双键的反应速率相近。
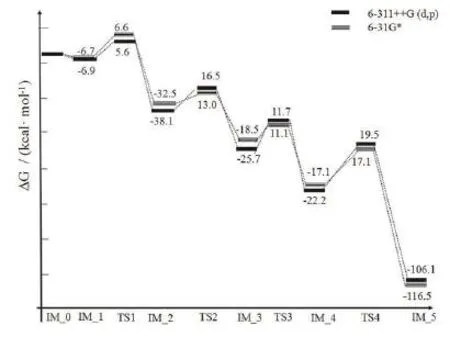
图5 臭氧和苯并噻吩的反应能级图(Path 1)Fig. 5 Energetic profile of the mechanism of ozone and benzothiophene ( Path 1 )
另外,在图6中给出了臭氧氧化苯并噻吩生成亚砜并且进一步氧化生成砜的机理(Path 2)。首先臭氧中的O1进攻苯并噻吩上的硫原子S1,通过过渡态TS5氧化生成亚砜。通过分析,O1-O2键断裂,由原来的1.264 Å 伸长为 2.379 Å,新键S1-O1键生成,由原来的2.930Å 缩短为 1.521 Å。接着另一分子的臭氧上的氧原子O4进攻亚砜,O4-O5断裂,由原来的1.256 Å 伸长为 2.746 Å。新键S1-O4生成,由原来的2.866 Å缩短为1.468 Å,经过过渡态TS6氧化生成砜。从整体势能面图7来看,第一步的活化能最大,为23.7 kcal·mol-1。因此整个反应的控制步骤为臭氧分子中氧分子进攻苯并噻吩上硫原子氧化生成亚砜的过程。但比Path 1的速控步高出6.6 kcal·mol-1,由此表明臭氧氧化不饱和键的反应速率高于其氧化饱和键的反应速率,臭氧氧化苯并噻吩遵循Path 1的可能性较大。
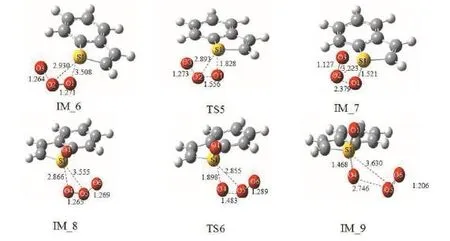
图6 臭氧和苯并噻吩反应各个驻点的几何构型(Path 2)Fig. 6 The geometries of each static point of ozone and benzothiophene's reaction (Path 2)
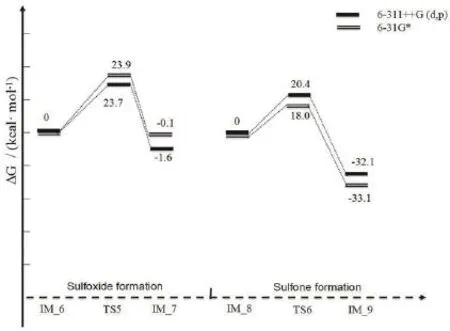
图7 臭氧和苯并噻吩的反应能级图(Path 2)Fig. 7 Energetic profile of the mechanism of ozone and benzothiophene ( Path 2 )
3.2 臭氧和苯并噻吩类衍生物反应
臭氧和苯并噻吩类衍生物二苯并噻吩及 4,6-二甲基苯并噻吩的反应机理与Path 2相类似,优化得出的几何结构分别如图8和图9所示。经过对分子的结构和相互作用的分析,不难发现,两个反应的机理非常相似:二苯并噻吩的噻吩环上的硫原子S1被臭氧中的氧分子进攻,经过过渡态TS7氧化生成亚砜,接着另一分子臭氧进一步进攻亚砜上的S1原子,经过 TS8氧化生成了砜。4,6-二甲基苯并噻吩与臭氧的反应也遵循此规律。从整体势能面图10来看,二苯并噻吩与臭氧反应的速控步所需要的活化能垒是23.2 kcal·mol-1,比臭氧氧化4,6-二甲基苯并噻吩及苯并噻吩反应的速控步所需活化能分别高出2.1 kcal·mol-1及6.1 kcal·mol-1。因此可得出臭氧和苯并噻吩及其衍生物反应的活性顺序为:苯并噻吩> 4,6-二甲基苯并噻吩 >二苯并噻吩。研究结果和文献中的实验现象一致[17],证明了该机理的合理性。另外,有文献报道苯并噻吩、二苯并噻吩和 4,6-二甲基苯并噻吩上硫原子电子云密度分别为 5.739、5.758和5.760[25]。 说明臭氧在氧化苯并噻吩及其衍生物的反应速率不遵循硫原子电子云密度大小的顺序。原因为苯并噻吩臭氧化机理区别于二苯并噻吩及其衍生物的臭氧化机理,根据反应能垒大小可知,臭氧在氧化不饱和键的反应速率高于氧化饱和键的反应速率。同时臭氧在氧化二苯并噻吩及 4,6-二甲基苯并噻吩时,4,6-二甲基苯并噻吩的氧化速率高于二苯并噻吩的氧化速率,这是由于这两种有机硫化物的硫原子电子云密度的差异所造成的。
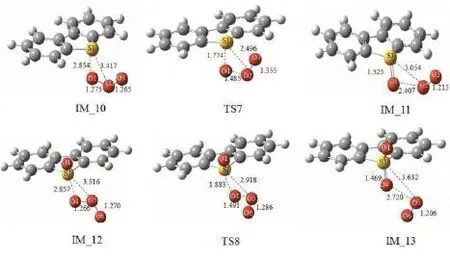
图8 臭氧和二苯并噻吩的反应各个驻点的几何构型Fig. 8 Ozone and dibenzothiophene geometries of each static point
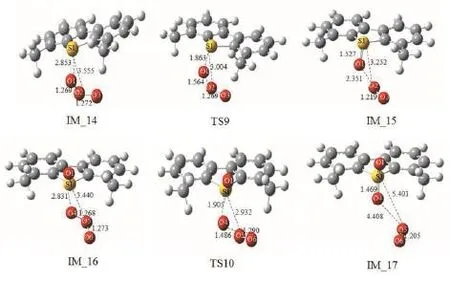
图9 臭氧和4,6-二甲基苯并噻吩的反应各个驻点的几何构型Fig. 9 Ozone and 4,6-dimethyldibenzothiophene geometries of each static point
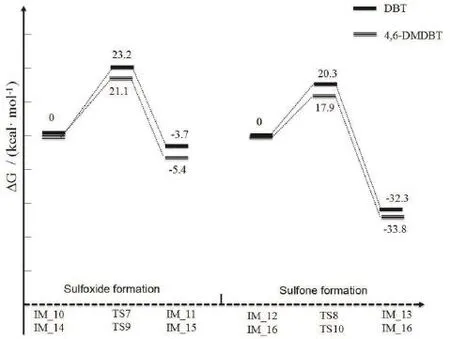
图10 臭氧和二苯并噻吩、4,6-二甲基苯并噻吩的反应能级图Fig. 10 Energetic profile of the mechanism of ozone and dibenzothiophene, 4,6-dimethyldibenzothiophene
4 结 论
采用密度泛函理论DFT/B3LYP方法,以6-31G (d, p)以及6-311++G(d, p)水平下预测了臭氧和苯并噻吩及其衍生物可能的反应路径。苯并噻吩与臭氧生成了稳定化合物2-巯基苯甲醛和二氧化碳。该反应的速控步为H2从C2原子处转移到S原子处,所需活化能为17.7 kcal·mol-1。此外,进一步研究了臭氧和苯并噻吩的衍生物(二苯并噻吩、4,6-二甲基苯并噻吩)的反应机理。实验结果表明,臭氧和二苯并噻吩、4,6-二甲基苯并噻吩反应机理相似,臭氧中的氧原子首先进攻硫原子氧化生成亚砜,进一步氧化成砜。由臭氧和苯并噻吩及其衍生物的活化能大小可得出其反应活性规律:苯并噻吩 > 4,6-二甲基苯并噻吩 > 二苯并噻吩。以上分析结果为臭氧氧化油品中有机硫化物的反应机理提供理论参考。
参考文献:
[1] PARK J, LEE H K, SOON A, et al. Thermally induced desulfurization: structural transformation of thiophene on the Si(100) surface[J]. The journal of physical chemistry C, 2013, 117(22): 11731-11737. DOI: 10.1021/jp306495k.
[2] KONG L Y, LI G, WANG X S, et al. Oxidative desulfurization of organic Sulfur in gasoline overAg/TS-1[J]. Energy & fuels, 2006, 20(3): 896-902. DOI: 10.1021/ef050252r.
[3] 李静, 曲久辉, 刘会娟, 等. 臭氧氧化水中黄腐酸过程中有机物的形态变化[J]. 中国科学 B辑: 化学, 2008,38(1): 67-71.
[4] ZENG X Y, WANG H L, DEYONKER N J, et al. Reaction mechanism of oxidative desulfurization of heterocyclic organic sulfides: a computational study[J]. Theoretical chemical accounts, 2014, 133: 1498. DOI: 10.1007/s00214-014-1498-1.
[5] 仇毅翔, 万明达, 陈先阳, 等. 金(I)配合物催化乙烯加氢的反应机理[J]. 物理化学学报, 2013, 29(2): 279-286. DOI: 10.3866/PKU.WHXB201212061.
[6] 袁俊辉, 高博, 汪文, 等. Y-Cu共掺杂ZnO电子结构与光学性质的第一性原理计算[J]. 物理化学学报, 2015,31(7): 1302-1308. DOI: 10.3866/PKU.WHXB201505081.
[7] 任清华, 沈晓燕. 铁催化芳基格氏试剂的联芳交叉偶联的反应机理[J]. 物理化学学报, 2015, 31(5): 852-858. DOI: 10.3866/PKU.WHXB201503026.
[8] WANG H L, HUANG D, ZHANG X, et al. Understanding the aqueous phase ozonolysis of isoprene: distinct product distribution and mechanism from the gas phase reaction[J]. Atmospheric chemistry and physics,2012, 12(15): 7187-7198. DOI: 10.5194/acp-12-7187-2012.
[9] ZHAO Y, ZHANG R X, WANG H, et al. Mechanism of atmospheric ozonolysis of sabinene: a DFT study[J]. Journal of molecular structure: THEOCHEM, 2010,942(1/3): 32-37. DOI: 10.1016/j.theochem.2009.11.029.
[10] KARAGULIAN F, LEA A S, DILBECK C W, et al. A new mechanism for ozonolysis of unsaturated organics on solids: phosphocholines on NaCl as a model for sea salt particles[J]. Physical chemistry chemical physics,2013, 10(48): 528-541. DOI: 10.1039/B712715D.
[11] ZHANG D, ZHANG R Y. Ozonolysis of α-pinene and β-pinene: kinetics and mechanism[J]. The journal of chemical physics, 2005, 122(11): 114308. DOI: 10.1063/ 1.1862616.
[12] SYROEZHKO A M, PROSKURYAKOV V A, BEGAK O Y. Mechanism of cyclohexane ozonolysis considering alternative pathways[J]. Russian journal of applied chemistry, 2002, 75(9): 1448-1452. DOI: 10.1023/A: 1022233129981.
[13] ANGLADA J M, CREHUET R, BOFILL J M. The ozonolysis of ethylene: a theoretical study on the gas-phase reaction mechanism[J]. Chemistry-a european journal, 1999, 5(6): 1809-1822. DOI: 10.1002/(SICI)1521-3765(19990604)5:6<1809::AID-CHEM1809>3.0. CO;2-N.
[14] GELETNEKY C, BERGER S. The mechanism of ozonolysis revisited by17O-NMR spectroscopy[J]. European Journal of organic chemistry, 1998, 1998(8): 1625-1627. DOI: 10.1002/(SICI)1099-0690(199808)1998:8<1625:: AID-EJOC1625>3.0.CO;2-L.
[15] DENINNO M P. “Anomalous” ozonolysis of cyclic allylic alcohols: mechanism and synthetic utility[J]. Journal of the American chemical society, 1995, 117(39): 9927-9928.
[16] CRIEGEE R. Mechanism of ozonolysis[J]. Angewandte chemie international edition, 1975, 14(11): 745-752.
[17] 张伟. 分子氧氧化法深度脱除燃油中有机硫研究[D].广州: 华南理工大学, 2014: 1-138.
[18] JIANG W, ZHU W S, CHANG Y H, et al. Oxidation of aromatic sulfur compounds catalyzed by organic hexacyanoferrates in ionic liquids with a low concentration of H2O2as an oxidant[J]. Energy & fuels,2014, 28(4): 2754-2760. DOI: 10.1021/ef500082y.
[19] MURATA S, MURATA K, KIDENA K, et al. A novel oxidative desulfurization system for diesel fuels with molecular oxygen in the presence of cobalt catalysts and aldehydes[J]. Energy & fuels, 2004, 18(1): 116-121. DOI: 10.1021/ef034001z.
[20] DEHU C, MEYERS F, BREDAS J L. Donor-acceptor diphenylacetylenes: geometric structure, electronic structure,and second-order nonlinear optical properties[J]. Journal of the American chemical society, 1993, 115(14): 6198-6206. DOI: 10.1021/ja00067a039.
[21] LEE C, YANG W T, PARR R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Physical review b,1988, 37(2): 785-789. DOI: 10.1103/PhysRevB.37.785.
[22] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09, revision D.01. Gaussian Inc. Wallingford,CT, 2013.
[23] KUGEL R W, AULT B S. Infrared matrix isolation and theoretical studies of reactions of ozone with bicyclic alkenes: α-pinene, norbornene, and norbornadiene[J]. The journal of physical chemistry a, 2015, 119(2): 312-322. DOI: 10.1021/jp510883k.
[24] 李来才. 乙烯与臭氧反应机理的理论研究[J]. 四川师范大学学报(自然科学版), 2003, 26(4): 388-392. DOI: 10.3969/j.issn.1001-8395.2003.04.017.
[25] OTSUKI S, NONAKA T, TAKASHIMA N, et al. Oxidative desulfurization of light gas oil and vacuum gas oil by oxidation and solvent extraction[J]. Energy & fuels, 2000, 14(6): 1232-1239. DOI: 10.1021/ef000096i.
Quantum Chemistry Study on the Reaction Mechanism of Ozone Oxidation of Benzothiophene and Its Derivatives
JIANG Min-jie, WANG Han-lu, LIANG Wei, ZHOU Jian-Min
(Guangdong University of Petrochemical Technology, College of Chemistry Engineering, Maoming 525000, Guangdong, China)
Abstract:A theoretical study on the geometries, reaction paths, as well as the frontier molecular orbitals of the ozone oxidation of benzothiophene and its derivatives was conducted by using the density functional theory B3LYP method at 6-31G* and 6-311++G(d, p) levels. Firstly, ozone attacked on the double bond of benzothiophene (BT). The highest occupied molecular orbital (HOMO) of the ozone and the lowest unoccupied molecular orbital (LUMO) of benzothiophene are symmetrical matched each other to generate a complex. Finally, 2-mercapto-benzoic acid was produced by breaking the double five-membered ring. Furthermore, the ozonation of dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) are calculated. The sulfides were oxidized to sulfoxide, and further oxidized to sulfone. Based on the activation energies, the reactivity of benzothiophene and its derivatives to ozone are in the following order: BT > 4,6-DMDBT > DBT.
Key words:ozone oxidation; benzothiophene; reaction mechanism; oxidation activity; DFT
中图分类号:TK421;X5
文献标志码:A
doi:10.3969/j.issn.2095-560X.2016.02.0010
文章编号:2095-560X(2016)02-0139-07
* 收稿日期:2015-12-20
修订日期:2016-03-10
基金项目:国家自然科学青年基金(21403038);广东省自然科学基金(2015A030313892);广东省高等学校优秀青年教师培养计划(YQ2015116);广东省大学生科技创新培育专项资金项目“攀登计划”(pdjh2015b0367);茂名市科技计划(2014084)
通信作者:†王寒露,E-mail:18630332@qq.com
作者简介:
蒋敏结(1994-),女,主要从事臭氧的量子化学研究。
王寒露(1982-),女,博士,讲师,主要从事油品清洁化技术利用与开发。
