利用CRISPR/Cas 9技术构建大肠杆菌aroA基因的敲除系统及其初步应用
2016-07-11余深翼赵金荣郑玲红朱二鹏周五朵吴宝成
余深翼,赵金荣,郑玲红,朱二鹏,周五朵,吴宝成
(福建农林大学动物科学学院预防兽医实验室,福州 350002)
利用CRISPR/Cas 9技术构建大肠杆菌aroA基因的敲除系统及其初步应用
余深翼,赵金荣,郑玲红,朱二鹏,周五朵,吴宝成*
(福建农林大学动物科学学院预防兽医实验室,福州 350002)
摘要:旨在利用CRISPR/Cas 9新型基因编辑技术构建大肠杆菌aroA基因的敲除系统,并分析其对不同大肠杆菌aroA基因敲除、修复的差异。首先构建靶向aroA基因的CRISPR/Cas 9载体;随后人工设计同源修复供体基因序列,并亚克隆到CRISPR/Cas 9载体中,构建成完整的CRISPR/Cas 9基因敲除体系;将该系统分别应用到大肠杆菌DH10B、DH5α和JM109细胞中,PCR鉴定筛选到的阳性菌株,并回收其扩增条带进行克隆、测序。CRISPR/Cas 9载体的酶切与测序结果均正确,表明载体构建成功;PCR鉴定结果显示,该系统对三种大肠杆菌均能进行aroA基因的有效敲除,敲除效率为46%~58%;测序结果进一步证实目的基因敲除成功。本试验成功构建大肠杆菌aroA基因CRISPR/Cas 9敲除系统,为进一步研究致病菌aroA基因功能及开发减毒大肠杆菌疫苗提供新型、有效的基因敲除工具。
关键词:CRISPR/Cas 9系统;大肠杆菌;aroA基因;敲除;同源修复
aroA基因负责编码5-烯醇丙酮酰莽草酸-3-磷酸合成酶(5-enolpyruvylshikimate-3-phosphate synthase,EPSPS),该酶可催化芳香族氨基酸的合成,是细菌芳香族氨基酸代谢中的重要酶类。芳香族氨基酸生物合成途径是革兰阴性菌和阳性菌共有的途径,而哺乳动物不具备该合成途径[1-2]。aroA基因最早在大肠杆菌中被发现,随后在其他细菌中也陆续被发现。研究人员发现,aroA基因的缺失导致EPSPS不能正常合成,细菌在无此合成途径的哺乳动物宿主内无法获得该酶,从而影响细菌的正常生长,同时也降低了相应的毒力[3]。aroA基因缺失的营养缺陷型菌株的选育,为新型减毒活疫苗的研制奠定了基础。早在1981年,S.K.Hoiseth等就对伤寒沙门菌的aroA基因做了敲除研究,发现缺失aroA基因的菌株,其毒力明显降低[3]。L.H.Garside等利用同源重组技术,对胸膜肺炎放线杆菌的aroA基因进行敲除,并成功筛选出了缺失菌株[4]。T.Kim等在体外表达猪圆环病毒的CAP基因,并利用aroA基因缺失的支气管波氏杆菌作为活疫苗载体,构建重组细菌疫苗,免疫猪能够很好地诱导产生中和抗体[5]。
成簇、规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)是许多细菌和古细菌抵御外来噬菌体入侵的一种免疫机制,最早被发现于大肠杆菌基因中[6-7]。CRISPR通常由前导区(leader)、间隔区(spacers)以及重复序列区(repeats)三部分组成[8-9]。原间隔序列旁侧存在一个保守的短序列,称为PAM (protospacer-adjacent motif),该序列作为Cas 蛋白对原间隔序列识别的一个序列信号。在CRISPR位点侧旁通常包含一组保守的CRISPR相关基因(CRISPR-associated genes,Casgenes),可以编码具有核酸酶和解旋酶活性的Cas蛋白[10-11]。Cas蛋白与CRISPR转录出来的RNA共同组成蛋白质核酸复合物,并发挥免疫功能[12]。Cas 9 是Cas蛋白家族中的一员,与CRISPR共同构成II型CRISPR/Cas系统,行使其核酸内切酶功能,对靶点DNA序列进行特异性切割,从而实现对基因组的精确编辑[13-14]。原核细胞大多缺乏非同源末端连接修复系统,需对切割的基因组DNA进行人工同源重组修复,进而实现原核基因组精确的敲除、修饰等[15]。
本研究基于CRISPR/Cas 9基因编辑技术,构建含有aroA基因靶向RNA(sgRNA)及用于同源修复的Donor序列的质粒,分析其对不同大肠杆菌aroA基因的敲除效率,并在较短的时间内筛选出aroA基因缺失菌株,为研发新型疫苗载体和深入探析致病菌aroA基因功能奠定基础,同时也为研究其他动物病原体重要基因功能及其作用机制提供重要平台。
1材料与方法
1.1菌株和质粒
pEwt-Cas 9 和psgRNA-GFP质粒均由Biomics Biotech公司提供,构建psgRNA-GFP质粒时,插入不含有BsaⅠ氨苄抗性基因(其氨苄抗性基因序列与pX458质粒的氨苄抗性基因序列一致)。pUC 57质粒由本实验室保存,DH10B、DH5α和JM109感受态细胞均由本实验室制备并保存。 sgRNA 启动子为组成型启动子J23100,Cas 9启动子为化脓链球菌spcas9天然启动子。
1.2主要试剂
胶回收试剂盒(Gel Extraction Kit)和质粒小提试剂盒(Plasmid Mini Kit)均购于OMEGA公司;DNA Marker、Premix ExTaqTMMix Version 2.0和TIANamp Bacteria DNA Kit均购于TaKaRa公司;氯霉素(chloromycetin)和氨苄青霉素(ampicillin)均购于上海生工公司。
1.3sgRNA的设计和相关引物的合成
aroA基因sgRNA由百奥迈科生物技术有限公司设计。根据DH10B菌株的aroA基因序列(GenBank序列号CP000948.1),在aroA基因CDS区的+41位置设计靶向sgRNA,并命名为aroAsgRNA,分别在sgRNA的上下游5′端添加BsaⅠ黏性末端。根据aroA编码基因的上下游序列设计左、右同源臂引物,分别为aroA L F/R和aroA R F/R。此外,还设计了sgRNA及Donor质粒构建过程中需要的其他引物aroA Aml F/R、aroA LRF-M13F/R以及aroA基因鉴定引物aroAP F/R。各引物和sgRNA寡核苷酸序列及目的片段大小见表1。所有的引物和寡核苷酸链均由百奥迈科生物技术有限公司合成。
1.4sgRNA载体的构建
取等量的sgRNA上下游引物混合(终浓度为10 μmol·L-1),加入退火Buffer进行退火处理。退火条件:95 ℃ 10 min,慢慢降温到25 ℃。取BsaⅠ酶切后的psgRNA-GFP质粒1 μL与稀释后的双链sgRNA退火产物 4 μL,22 ℃下连接2 h。将连接产物转化到DH5α 感受态细胞中,用带氨苄青霉素抗性的平板筛选,挑取单克隆进行摇菌,小提质粒,并做酶切和测序鉴定。把构建好的质粒命名为psgRNA-GFP-aroAsgRNA。
表1sgRNA和引物及其目的片段大小
Table 1Sequences and product length of sgRNA and primers

sgRNA/引物sgRNA/Primers序列Oligonucleotidesequences片段/bpLengtharoAsgRNAFaroAsgRNAR5'-GATCCCAACCCATCGCTCGTGTCGAT-3'(BsaⅠ)5'-AAACATCGACACGAGCGATGGGTTGG-3'(BsaⅠ)20aroALFaroALR5'-CGCGGATCCACGAAACGCCAGACTTCGG-3'(BamHⅠ1011282-1011300)5'-CCCAAGCTTGTTCGAACTCAACCATGA-3'(HindⅢ1011861-1011878)600aroARFaroARR5'-CGCGGATCCGCCGAGATCGCGACATACAA-3'(BamHⅠ 1013091-1013110)5'-CCCAAGCTTACTTGATTGCCGCTGTCCAT-3'(HindⅢ 1013592-1013612)520aroAAmlFaroAAmlR5'-CCGGAATTCACGAAACGCCAGACTTCGG-3'(EcoRⅠ)5'-CTAGTCTAGACGTTCGAACTCAACCATGAAGT-3'(XbaⅠ)610aroALRF-M13FaroALRF-M13R5'-GTTGTAAAACGACGGCCAGT-3'5'-CGCCATATGTTCACACAGGAAACAGCTATGA-3'(NdeⅠ)1250aroAPFaroAPR5'-CCATCGACGAAACGCCAGAC-3'(1011276-1011295)5'-TGTTACCCAATGCTTTGGCGA-3'(1013653-1013673)2400/1200
1.5aroA基因同源臂(Donor)载体的构建
以DH10B细菌基因组DNA为模版,aroA L F/R和aroA R F/R为引物,PCR扩增左右同源臂(以下称Donor)序列,即Donor aroA L和aroA R。采用25 μL体系:DNA模板2 μL,PremixTaqMix 12.5 μL,aroA L(或R)上下游引物各1 μL,ddH2O 8.5 μL。反应条件为95 ℃预变性5 min,之后95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s进行30个循环;最后72 ℃延伸7 min。取PCR扩增产物,进行1%琼脂糖凝胶电泳鉴定。
分别对回收的PCR产物和pUC 57质粒进行BamHⅠ+Hind Ⅲ双酶切(37 ℃,2 h),22 ℃连接2 h后转化DH5α感受态细胞,筛选并挑取单克隆菌落,摇菌后小提质粒,并作酶切和测序鉴定。把构建好的质粒命名为pUC57-aroA L 和pUC57-aroA R。
以pUC57-aroA L为模板,aroA Aml F/R为引物,PCR扩增出两端含EcoRⅠ和XbaⅠ酶切位点的aroAL基因序列。分别对回收的aroAL基因和pUC57-aroA R质粒进行EcoRⅠ+XbaⅠ双酶切、连接,并转化到DH5α感受态细胞,筛选并挑取单克隆菌落,摇菌后小提质粒,并作酶切和测序鉴定。把构建好的质粒命名为pUC57-aroA L R(Donor)。
1.6含sgRNA及aroA基因同源臂载体的构建
以构建好的质粒pUC57-aroA L R为模板,aroALRF-M13F 和aroALRF-M13R为引物,PCR扩增出含有EcoRⅠ和NdeⅠ的aroAL R 基因,分别对回收的aroAL R基因和psgRNA-GFP-aroA sgRNA质粒进行EcoRⅠ+NdeⅠ双酶切、连接,并转化到DH5α感受态细胞,筛选并挑取单克隆菌落,摇菌后小提质粒,并作酶切鉴定和测序鉴定。质粒命名为psgRNA-GFP-aroA sgRNA-Donor。
1.7感受态细胞制备及细菌的转化
DH5α、DH10B和JM109三种大肠杆菌aroA基因相似性高达98%,故推测本试验构建的针对aroA基因的CRISPR/Cas 9敲除系统可能适用于这三种菌。参考《分子克隆实验指南》,按照CaCl2法分别制备DH5α、DH10B、JM109的化学感受态细胞,把构建好的psgRNA-GFP-aroAsgRNA-Donor质粒和pEwt-Cas 9质粒分别先后转化到DH5α、DH10B和JM109感受态细胞中。
1.8稳定敲除aroA基因的DH5α、DH10B、JM109菌株的筛选
分别用带氯霉素和氨苄青霉素抗性的平板筛选同时含有psgRNA-GFP-aroAsgRNA-Donor质粒和pEwt-Cas 9质粒的菌株,用针对aroA基因的特异性引物aroA P F和aroA P R进行菌液PCR检测。对PCR检测敲除成功的菌,回收其PCR产物进行TA克隆,并送InvitrogenTM生物公司测序,将测序结果和原基因进行比对,检测aroA基因是否敲除成功。
2结果
2.1sgRNA载体的构建
将退火后形成的双链sgRNA定向亚克隆到psgRNA-GFP(氨苄抗性基因序列中不含有BsaⅠ酶切位点)质粒中,将构建好的质粒载体psgRNA-GFP-aroA sgRNA送检测序,测序结果正确,可用于下一步试验。sgRNA载体构建图谱如图1所示。
2.2aroA基因同源臂载体的构建
分别以aroA L F/R和aroA R F/R为引物扩增aroA基因左右两端同源臂:aroAL(600 bp)和aroAR(520 bp)。琼脂糖凝胶电泳检测结果(如图2A)显示,PCR扩增产物均与预期的条带大小相符。
分别对左右同源臂重组质粒pUC57-aroA L(3 330 bp)和pUC57-aroA R(3 220 bp)进行酶切鉴定,结果如图2A所示,重组质粒经过BamHⅠ+Hind Ⅲ双酶切后可见2 700和600 bp (520 bp)的两条特异性条带,均与预期大小一致。阳性重组质粒的测序结果进一步证实序列完全正确。
以aroA Aml F和aroA Aml R为引物,pUC57-aroA L为模板,PCR扩增出paroAL(610 bp)序列,结果与预期大小一致(图2B)。对重组的质粒pUC57-aroA L R(3 850 bp)进行酶切鉴定,结果如图2B所示,重组质粒经过EcoRⅠ+XbaⅠ双酶切后可见2 700和1 150 bp的两条特异性条带,均与预期大小一致。阳性重组质粒的测序结果显示完全正确。
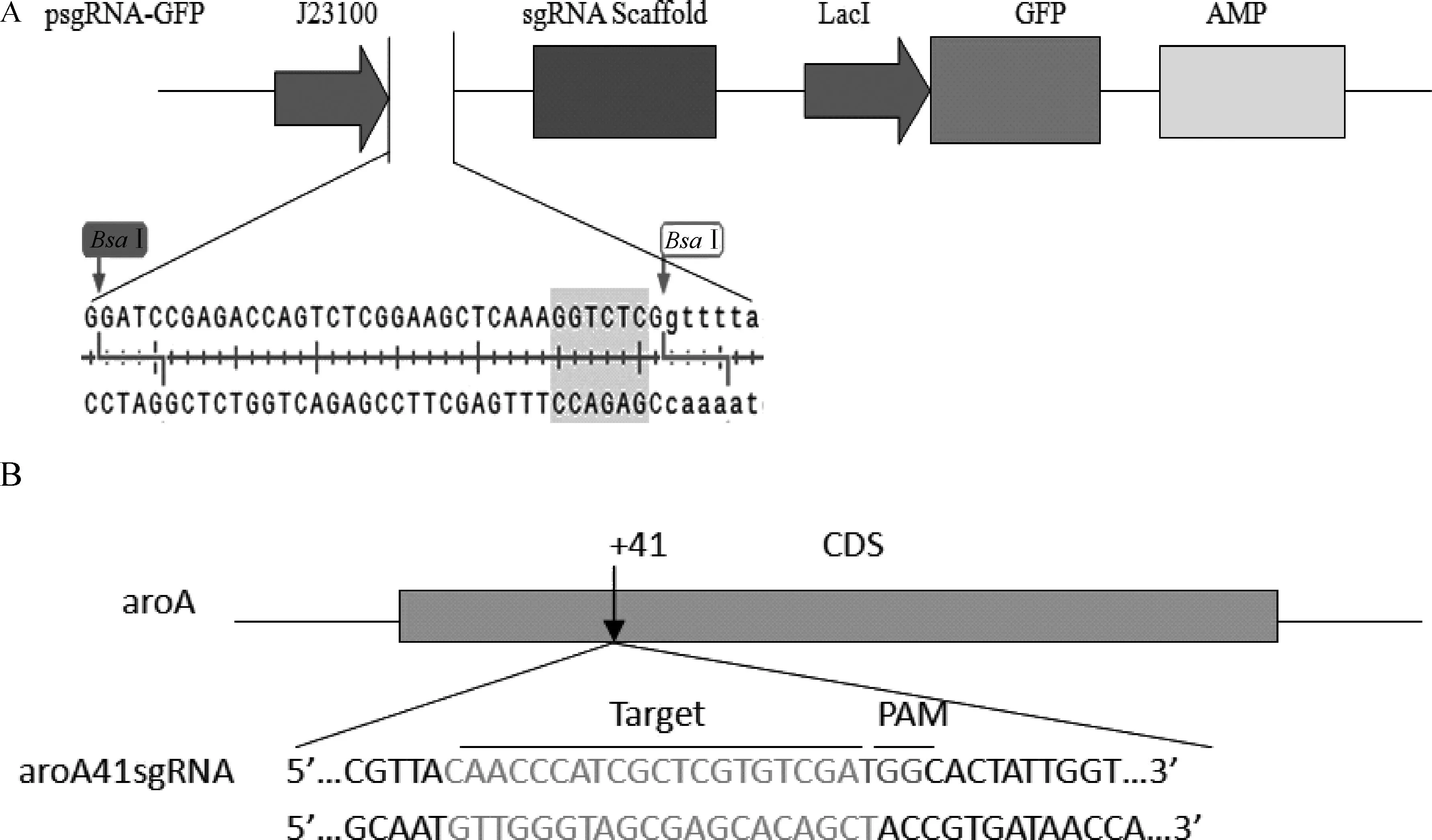
A.质粒的部分图谱以及BsaⅠ酶切位点位置;B.aroA基因靶向sgRNA的插入位置及其周边DNA序列A.Partial map of psgRNA-GFP plasmid and location of Bsa Ⅰ restriction site;B.Insertion position and flanking sequences of aroA gene-targeted sgRNA图1 靶向aroA基因的psgRNA-GFP-aroA sgRNA质粒构建图谱Fig.1 Schematic representation of psgRNA-GFP-aroA sgRNA construction
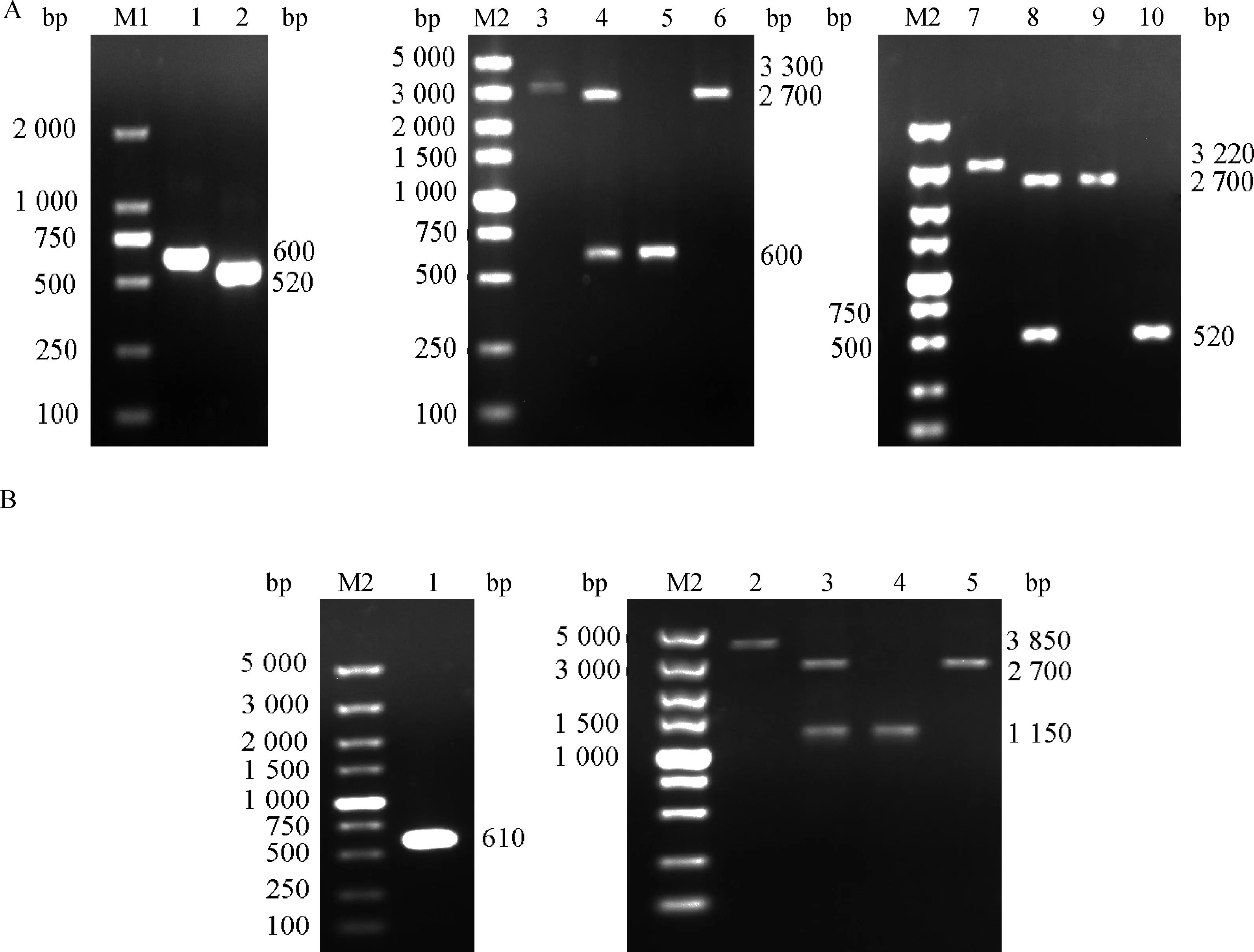
A.aroA L(左)和aroA R(右)同源臂载体构建:M1、M2.DNA相对分子质量标准;1.aroA L扩增结果;2.aroA R扩增结果;3.重组质粒pUC57-aroA L的BamHⅠ单酶切产物;4.pUC57-aroA L的BamHⅠ+Hind Ⅲ双酶切产物;5.aroA L基因对照;6.线性化的pUC57;7.重组质粒pUC57-aroA R的BamHⅠ单酶切产物;8.pUC57-aroA R的BamHⅠ+HindⅢ双酶切产物;9.线性化的pUC57;10.aroA R基因对照。B.aroA L R同源臂载体构建:M2.DNA相对分子质量标准;1.paroA L扩增产物;2.重组质粒pUC57-aroA L R 的EcoRⅠ单酶切产物;3.pUC57-aroA L R的EcoRⅠ+XbaⅠ双酶切产物;4.aroA L R基因对照;5.线性化的pUC57A.Construction of the recombinant plasmid pUC57-aroA L and pUC57-aroA R:M1.DL2000 marker;M2.DL10000 marker;1.PCR product of aroA L;2.PCR product of aroA R;3.Single digestion of pUC57-aroA L by BamH Ⅰ;4.Double digestion of pUC57-aroA L by BamHⅠ+Hind Ⅲ;5.aroA L control;6.Linearized pUC57;7.Single digestion of pUC57-aroA R by BamHⅠ;8.Double digestion of pUC57-aroA R by BamHⅠ+HindⅢ;9.Linearized pUC57;10.aroA R control.B.Construction of the recombinant plasmid pUC57-aroA L R:M2.DL5000 marker;1.PCR product of paroA L;2.Single digestion of pUC57-aroA L R by EcoRⅠ;3.Double digestion of pUC57-aroA L R by EcoRⅠ+XbaⅠ;4.aroA L R control;5.Linearized pUC57图2 aroA基因同源臂载体构建的鉴定Fig.2 PCR and restriction enzyme digestion analysis of the recombinant plasmid pUC57-aroA L R (Donor)
2.3psgRNA-GFP-aroAsgRNA-Donor载体的构建
以pUC57-aroA L R为模板,aroALR-M13F和aroALR-M13R为引物,PCR扩增出aroAL R(1 200 bp)基因片段。琼脂糖凝胶电泳检测结果(图3)显示与预期的条带一致。
对构建好的psgRNA-GFP-aroAsgRNA-Donor(4 900 bp)质粒用EcoRⅠ+NdeⅠ内切酶做酶切鉴定,结果如图3所示,重组质粒经过EcoRⅠ+NdeⅠ双酶切后可见3 750和1 150 bp的两条特异性条带,均与预期大小一致。将重组好的质粒送测序鉴定,结果与原序列一致。psgRNA-GFP-aroAsgRNA-Donor载体的构建过程如图4A所示。
2.4对不同大肠杆菌aroA基因敲除效率的检测
用以上构建好的CRISPR/Cas 9系统对DH5α、DH10B、JM109菌株分别进行aroA基因的敲除,aroA基因敲除模式如图4B所示。用横跨aroA基因同源臂的鉴定引物aroA P F/R扩增目的条带,鉴定aroA基因的敲除情况,结果如图5所示,aroA基因没被敲除修复的菌,PCR显示为2.4 kb的条带,反之出现1.2 kb的目的条带。该系统对DH5α、DH10B、JM109菌株的敲除效率分别为54%、46%和58%。并分别对以上敲除后形成小片段扩增产物(1.2 kb)进行TA克隆,送检测序。测序结果与设计的Donor序列是一致的,表明被Cas 9敲除后的aroA基因在人工设计的Donor序列下发生了同源修复。以上结果均表明,本研究构建的CRISPR/Cas 9系统在人工同源修复下,可对大肠杆菌aroA基因进行稳定的敲除。
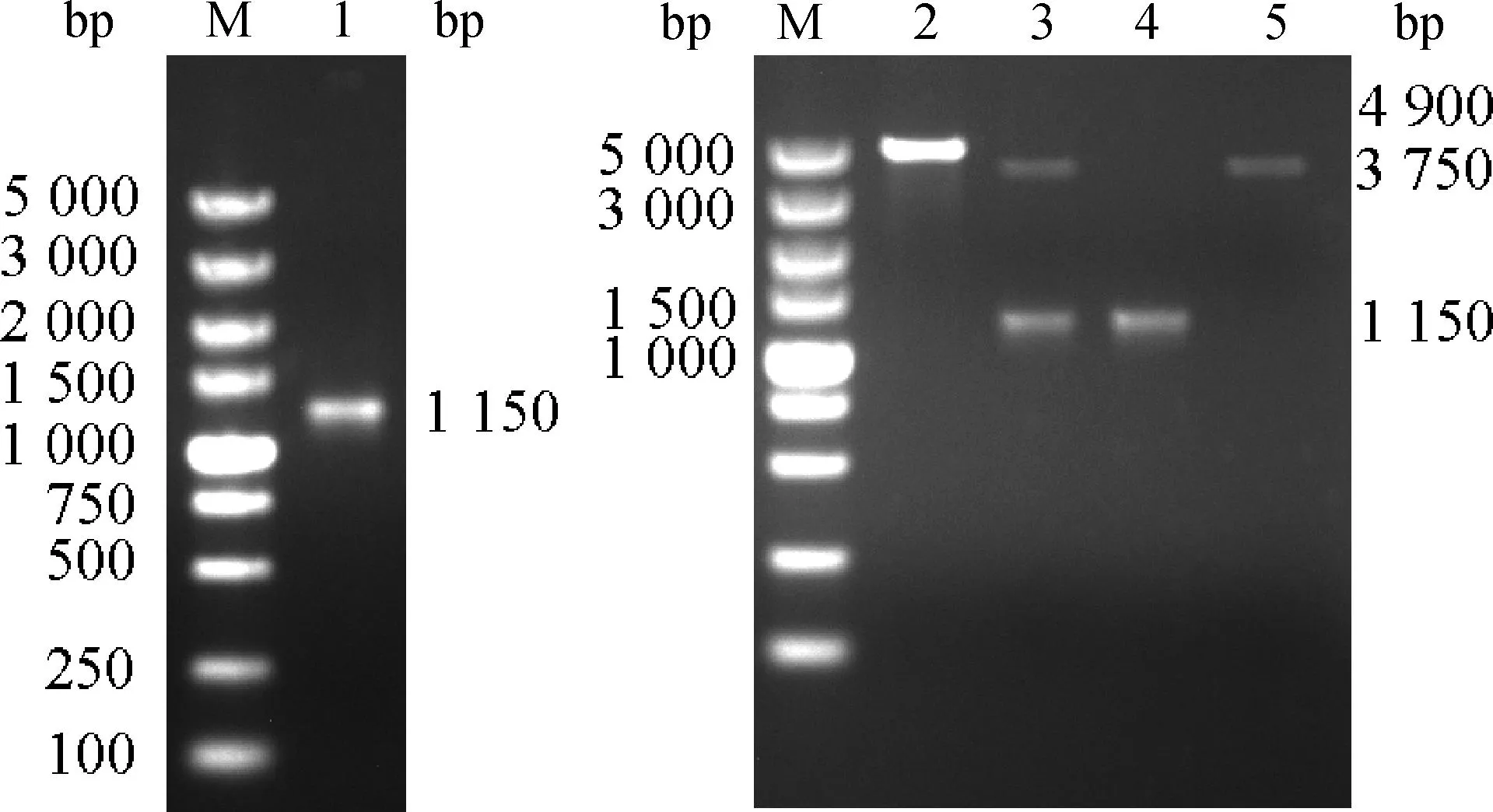
M.DNA相对分子质量标准;1.aroA L R基因扩增产物;2.重组质粒psgRNA-GFP-aroAsgRNA-aroALR的 EcoRⅠ单酶切产物;3.psgRNA-GFP-aroAsgRNA-aroALR的EcoRⅠ+NdeⅠ双酶切产物;4.aroA L R基因对照;5.线性化的psgRNA-GFP-aroAsgRNAM.DL5000 marker;1.PCR product of aroA L R;2.Single digestion of psgRNA-GFP-aroAsgRNA-aroALR by EcoRⅠ;3.Double digestion of psgRNA-GFP-aroAsgRNA-aroALR by EcoRⅠ+ NdeⅠ;4.aroA L R control;5.Linearized psgRNA-GFP-aroAsgRNA图3 psgRNA-GFP-aroAsgRNA-Donor载体构建检测Fig.3 PCR and restriction enzyme digestion analysis of the recombinant plasmid
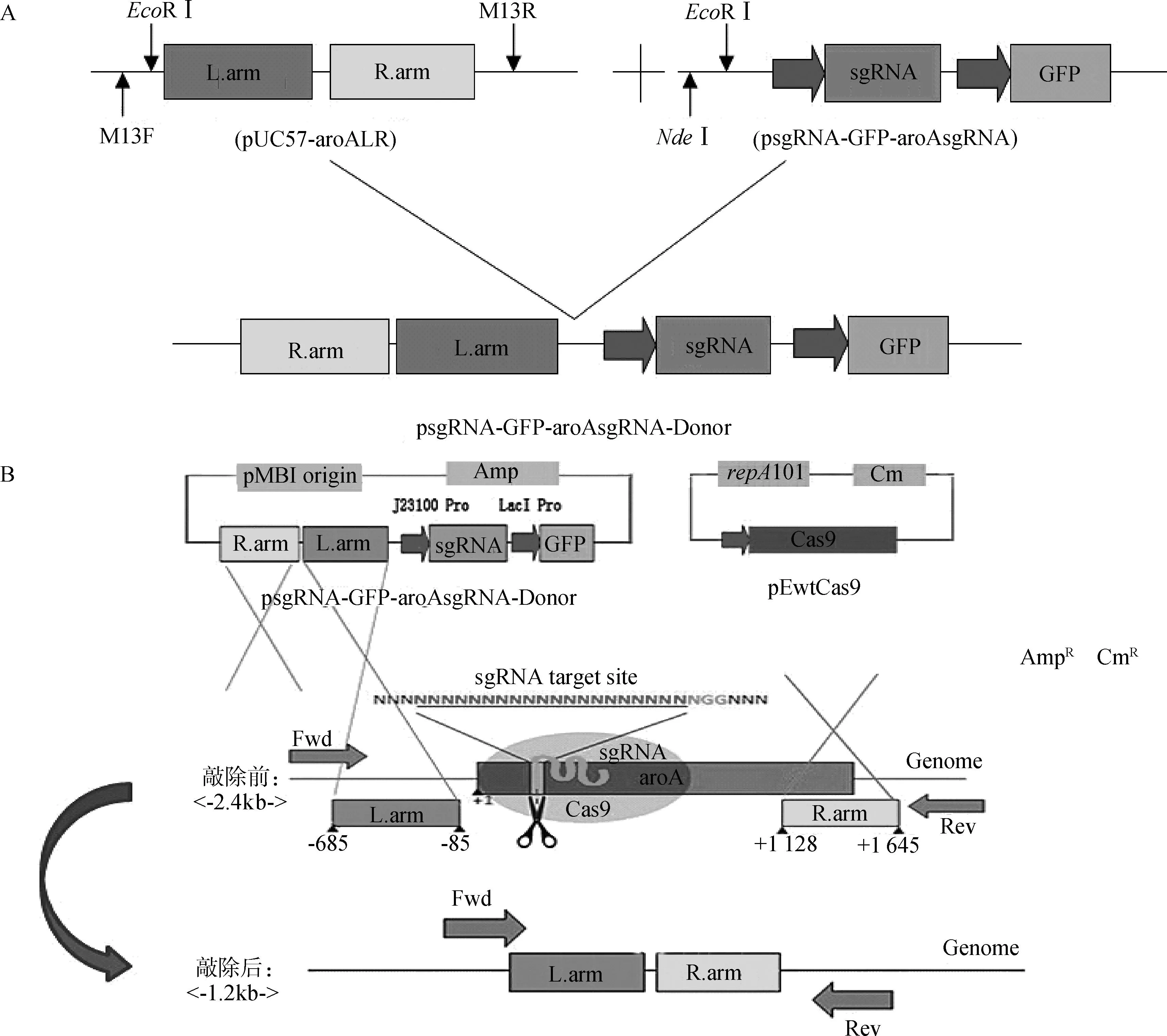
A.psgRNA-GFP-aroAsgRNA-Donor载体的构建过程;B.aroA基因的敲除模式A.Costruction of recombinant plasmid psgRNA-GFP-aroAsgRNA-Donor;B.Detailed diagram of continual genome editing with the two-plasmid CRISPR/Cas 9 system图4 aroA基因同源修复载体的构建及其敲除模式Fig.4 Schematic representation of psgRNA-GFP-aroAsgRNA-Donor construction knockout pattern
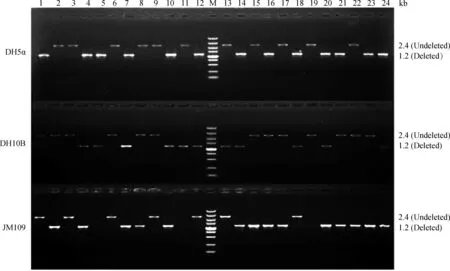
M.DNA相对分子质量标准;1~24.分别是24个双阳性单克隆菌落的PCR扩增结果,未敲除的PCR扩增产物大小为2.4 kb,敲除后的PCR扩增产物大小为1.2 kbM.DL5000 marker;1~24.24 monoclonal colonies for PCR amplification,the undeleted and deleted PCR products are 2.4 kb and 1.2 kb,respectively图5 aroA基因敲除后的PCR鉴定Fig.5 PCR and sequencing analysis of the post-knockout aroA gene
3讨论
CRISPR/Cas 9系统是研究人员从细菌和古细菌的免疫机制中发现的一种基因编辑技术,该技术与锌指核酸内切酶(ZFN)和类转录激活因子效应物核酸酶(TALEN)技术统称为三大人工核酸酶技术[16]。早期的ZFN技术和TALEN技术,都是通过DNA蛋白复合物对靶点进行特异性的识别和切割[17]。这两种技术不仅在构建相关质粒时费时费力,而且后期操作中对技术要求很高,因此在普通实验室很难普及,这也限制了此技术的广泛应用。CRISPR/Cas 9基因编辑技术对靶位点的识别,主要依赖于与靶标DNA互补配对的sgRNA。因此需要根据目的基因靶位点的PAM序列设计sgRNA,通常大小为20 bp[12]。表达的Cas 9蛋白可与sgRNA结合,形成蛋白质核酸复合物。该复合物识别特定的靶点DNA序列,发挥Cas 9核酸酶的作用,对靶位点进行精确的基因编辑[18]。质粒的构建和后续操作过程都相对简单、快捷且易于掌握。CRISPR/Cas 9基因编辑技术已然成为生命科学等研究领域的新宠[19],对动物及动物病原的基因改造研究越发重要。
致病菌是能引起许多动物发生传染病的病原,对其毒力基因进行基因组水平的改造,在选育强弱毒菌株等方面至关重要[20-21]。在CRISPR/Cas 9技术出现之前,主要应用同源重组对细菌进行基因组的编辑,应用比较成熟的同源重组系统有Red/Flp等[22],为解析基因功能和作用机制的研究做出了重要贡献。但单纯应用同源重组技术对基因组进行编辑的效率极低,且不同的物种间,构建好的同源重组质粒的基因编辑效率差异很大[23],对于不同的细菌,往往需要更换不同的条件和质粒。CRISPR/Cas 9系统的发现使得基因改造技术有了新的飞跃,该系统方便快捷,在原核生物尤其是细菌基因组改造中的应用研究已屡见不鲜[15-24]。研究人员在应用CRISPR/Cas 9技术编辑原核生物基因组时发现,目的基因被Cas 9剪切后,绝大多数不能修复自身断裂基因组,结果导致细菌死亡。而如果在此基础上存在同源修复的Donor序列时,断裂的基因双链则可发生自发性同源重组,且效率得到大幅提高[15]。因此,将CRISPR/Cas 9技术与同源重组技术结合起来,构建含Cas 9、sgRNA和Donor序列的重组载体,就能对原核基因进行有效准确编辑。CRISPR/Cas 9系统在发展的过程中有许多技术的改进,例如Y.Jiang等[24]在Cas 9质粒构建时引入了Red重组系统,使CRISPR/Cas 9针对一个细菌中的多个基因同时高效敲除,该系统的重要改进是Donor序列不需要克隆到载体里面,直接与质粒共转化,从而大大降低了试验难度。然而由于很多病原大肠杆菌的细胞壁结构跟大肠杆菌基因工程菌(例如DH5α、DH10B)细胞壁有很大的不同,转化效率较低,不适合直接大量共转化DNA片段作为供体,因此采用作者建立的CRISPR系统对这些难转化的病原大肠杆菌的基因敲除更有优势。本研究的结果表明,应用CRISPR系统可对大肠杆菌aroA基因进行快速的编辑,而细菌内残留质粒的消除还有待于进一步研究。Y.Jiang等[24]以温敏型质粒pKD46为基础,插入Cas 9基因,构建温敏型的Cas 9质粒,根据提供sgRNA和Donor的载体序列,设计一对靶向其自身的sgRNA序列。Cas 9蛋白完成目的基因敲除的同时,也会把提供sgRNA和Donor的载体切断。在完成敲除工作后,提高温度培养细菌,可使Cas 9质粒消失。
aroA基因是细菌在芳香烃生物合成途径中一个重要的酶[1],aroA基因缺失时,不能正常合成EPSPS[13],当该菌感染哺乳动物后,就无法合成相应的化合物,进而导致细菌生长抑制和毒力降低。早期有人用同源重组技术,对aroA基因进行改造,形成营养缺陷型的细菌,确实能降低致病菌的毒力[3]。Blast比对不同的大肠杆菌菌株aroA基因,可以发现其在不同菌株中序列非常保守,这就使构建一个不严格要求种间差异的通用敲除体系成为可能。本试验成功构建的CRISPR/Cas 9敲除系统含有特异靶向aroA基因sgRNA和Donor的基因元件,可对aroA基因进行定点的切割和同源重组修复,能对不同来源的大肠杆菌进行aroA基因敲除,结果显示其敲除效率没有太大差异。在该体系的应用过程中,使用的均不是致病菌,而是基因工程菌,所以接下来作者会进一步使用原有的体系,对临床中常见致病菌的aroA基因进行敲除,深入探索致病菌aroA基因缺失后的生物学功能变化以及其对哺乳动物致病力的影响。
4结论
构建了大肠杆菌aroA基因CRISPR/Cas 9敲除系统。该系统可对DH5α、DH10B、JM109三种大肠杆菌的aroA基因进行快速的敲除及修复,并可在较短的时间内筛选出aroA基因缺失菌株。为进一步研究致病菌aroA基因功能及开发减毒大肠杆菌疫苗提供新型、有效的基因敲除工具。
参考文献(References):
[1]BOOCOCK M R,COGGINS J R.Kinetics of 5-enolpyruvylshikimate-3-phosphate synthase inhibition by glyphosate[J].FEBSLett,1983,154(1):127-133.
[2]STEINRÜCKEN H C,AMRHEIN N.5-Enolpyruvylshikimate-3-phosphate synthase of Klebsiella pneumonia 2.Inhibition by glyphosate[N-(phosphonomethyl)glycine][J].EurJBiochem,1984,143(2):351-357.
[3]HOISETH S K,STOCKER B A.Aromatic-dependentSalmonellatyphimuriumare non-virulent and effective as live vaccines[J].Nature,1981,291(5812):238-239.
[4]GARSIDE L H,COLLINS M,LANGFORD P R,et al.Actinobacillus pleuropneumoniae serotype 1 carrying the defined aroA mutation is fully avirulent in the pig[J].ResVetSci,2002,72(2):163-167.
[5]KIM T,TOAN N T,SEO J,et al.BordetellabronchisepticaaroA mutant as a live vaccine vehicle for heterologous porcine circovirus type 2 major capsid protein expression[J].VetMicrobiol,2009,138(3-4):318-324.
[6]ISHINO Y,SHINAGAWA H,MAKINO K,et al.Nucleotide sequence of the iap gene,responsible for alkaline phosphatase isozyme conversion inEscherichiacoli,and identification of the gene product[J].JBacteriol,1987,169(12):5429-5433.
[7]BARRANGOU R,FREMAUX C,DEVEAU H,et al.CRISPR provides acquired resistance against viruses in prokaryotes[J].Science,2007,315(5819):1709-1712.
[8]JANSEN R,EMBDEN J D,GAASTRA W,et al.Identification of genes that are associated with DNA repeats in prokaryotes[J].MolMicrobiol,2002,43(6):1565-1575.
[9]JANSEN R,VAN EMBDEN J D,GAASTRA W,et al.Identification of a novel family of sequence repeats among prokaryotes[J].OMICS,2002,6(1):23-33.
[10]KUNIN V,SOREK R,HUGENHOLTZ P.Evolutionary conservation of sequence and secondary structures in CRISPR repeats[J].GenomeBiol,2007,8(4):R61.
[11]DEVEAU H,GARNEAU J E,MOINEAU S.CRISPR/Cas system and its role in phage-bacteria interactions[J].AnnuRevMicrobiol,2010,64:475-493.
[12]HWANG W Y,FU Y,REYON D,et al.Efficient genome editing in zebrafish using a CRISPR-Cas system[J].NatBiotechnol,2013,31(3):227-229.
[13]GARNEAU J E,DUPUIS M,VILLION M,et al.The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA[J].Nature,2010,468(7320):67-71.
[14]JINEK M,CHYLINSKI K,FONFARA I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J].Science,2012,337(6096):816-821.
[15]JIANG W,BIKARD D,COX D,et al.RNA-guided editing of bacterial genomes using CRISPR-Cas systems[J].NatBiotechnol,2013,31(3):233-239.
[16]MUSSOLINO C,CATHOMEN T.RNA guides genome engineering[J].NatBiotechnol,2013,31(3):208-209.
[17]GEURTS A M,COST G J,FREYVERT Y,et al.Knockout rats via embryo microinjection of zinc-finger nucleases[J].Science,2009,325(5939):433.
[18]MALI P,ESVELT K M,CHURCH G M.Cas9 as a versatile tool for engineering biology[J].NatMethods,2013,10(10):957-963.
[19]魏泽辉,贾存灵,张智英.CRISPR/Cas9系统在基因表达调控中的应用[J].畜牧兽医学报,2014,45(9):1387-1392.
WEI Z H,JIA C L,ZHANG Z Y.Application of CRISPR/Cas9 system for gene regulation[J].ActaVeterinariaetZootechnicaSinica,2014,45(9):1387-1392.(in Chinese)
[20]HARPER M,BOYCE J D,ADLER B.Pasteurellamultocidapathogenesis:125 years after Pasteur[J].FEMSMicrobiolLett,2006,265(1):1-10.
[21]SCHUBERT S,RAKIN A,KARCH H,et al.Prevalence of the “high-pathogenicity island”of Yersinia species amongEscherichiacolistrains that are pathogenic to humans[J].InfectImmun,1998,66(2):480-485.
[22]COPELAND N G,JENKINS N A,COURT D L.Recombineering:A powerful new tool for mouse functional genomics[J].NatRevGenet,2001,2(10):769-779.
[23]SHARAN S K,THOMASON L C,KUZNETSOV S G,et al.Recombineering:a homologous recombination-based method of genetic engineering[J].NatProtoc,2009,4(2):206-223.
[24]JIANG Y,CHEN B,DUAN C,et al.Multigene editing in theEscherichiacoligenome via the CRISPR-Cas9 system[J].ApplEnvironMicrobiol,2015,81(7):2506-2514.
(编辑白永平)
The Application of CRISPR/Cas 9 Technology for aroA Gene Knockout in Escherichia coli
YU Shen-yi,ZHAO Jin-rong,ZHENG Ling-hong,ZHU Er-peng,ZHOU Wu-duo,WU Bao-cheng*
(CollegeofAnimalScience,FujianAgricultureandForestryUniversity,Fuzhou350002,China)
Abstract:CRISPR/Cas 9 system is a novel gene editing technology.This study was aimed to construct CRISPR/Cas 9-based knockout system forE.coliaroAgene,and analyze knockout efficiencies in differentE.colis. In the present study,aroAgene-targeting short guide RNA (sgRNA) was designed and constructed together with Donor sequence for homologous repair,then this resulting vector and pEwt-Cas 9 vector co-constituted the CRISPR/Cas 9 system.Preliminary application was performed onE.coliDH10B,DH5α and JM109,respectively,and then knockout efficiencies ofaroAgene were analyzed by PCR amplification,TA cloning and subsequent DNA sequencing.The restriction enzyme digestion analysis and sequencing results showed that homologous repair vector was successfully constructed.PCR results indicated that the established CRISPR/Cas 9 system could be used foraroAgene knockout in multipleE.coliwith efficiency ranging from 46% to 50%.Sequencing results further confirmed the successful and accurate knockout ofaroAgene.In conclusion,CRISPR/Cas 9-based knockout system forE.coliaroAgene was successfully established,which would provide a novel and effective tool for functional studies onaroAgenes of other pathogenic microorganisms and further development of attenuated vaccine.
Key words:CRISPR/Cas 9 system;Escherichiacoli;aroAgene;knockout;homologous repair
doi:10.11843/j.issn.0366-6964.2016.04.016
收稿日期:2015-10-10
基金项目:福建省畜禽新发疫病诊断和防治技术研究(ky0040052)
作者简介:余深翼(1990-),男,福建福州人,硕士生,主要从事畜禽新发疫病诊断和防治技术研究,E-mail: yushenyi2014@163.com *通信作者:吴宝成,研究员,硕士生导师,E-mail: bc20020212@163.com
中图分类号:S852.612
文献标志码:A
文章编号:0366-6964(2016)04-0762-09
