芒果果实转录组数据组装及基因功能注释
2016-07-09武红霞许文天罗纯姚全胜王松标马小卫詹儒林
武红霞 许文天 罗纯 姚全胜 王松标 马小卫 詹儒林
摘 要 以红芒6号为试材,对生长发育各时期果皮与果肉样品提取RNA后等量混合进行转录组测序,共获得了68 419 722个reads,包含6 157 744 980个核苷酸序列信息,平均读长90 bp,序列信息全部登陆NCBI数据库(SRP035450)。对reads进行拼接,共获得124 002个Contig片段,进一步组装获得54 207个Unigene片段,平均长度为838 bp,阈值小于10-5的序列中共有42 515(78.43%)个unigenes被注释到公共蛋白数据库。其中,35 198个被注释到生物学过程、分子功能和细胞组分GO类别的44个功能组,14 619个Unigene匹配到25类COG功能组,23 741个Unigene被富集到128个KEGG代谢通路,包括代谢途径、次生代谢的生物合成、植物-病原物互作、植物激素信号转导、苯丙氨酸生物合成、淀粉和蔗糖代谢、类黄酮类化合物合成等。本研究将为进一步的芒果功能基因克隆、基因表达分析、分子标记开发等研究奠定基础。
关键词 芒果;转录组;功能注释
中图分类号 S682.2 文献标识码 A
芒果是著名的热带水果,素有“热带果王”之美誉。中国是主要的芒果生产国之一,2014年栽培面积14万hm2,产量130万t,主要分布在海南、广西、云南、四川、广东和福建等省(区)。前期研究主要集中于栽培生理研究,如果实成熟过程[1-4]、果实香气挥发物质[5-6]、抗氧化活性[7-8]、采后处理和果实品质研究[9-10],尽管芒果是中国热区农业产业的经济支柱,其基因组较小,但基因组信息相当缺乏。近年来,高通量转录组测序技术的不断发展和完善为开展不同生物功能基因组学研究提供了全新的思路和方法,转录组测序是基于下一代高通量测序技术建立的一种高效、快捷分子生物学研究手段,使科技工作者能在组学水平上研究基因组序列未知的非模式生物,从整体水平了解植物在特定阶段的基因功能及基因结构,更加便利地提示特定生物学过程的分子机制[11-13]。近年来,已广泛应用于杨梅[14]、苹果[15]、梨[16]、葡萄[17]、柑橘[18]等果树,芒果研究方面,Dautt-Castro等[19]用转录组测序技术研究了与 Kent芒成熟过程相关的基因,在青熟和成熟果实中,2 306个基因差异表达。Luria 等[20]用转录组测序揭示了热水处理对芒果影响的分子机制,基于基因表达谱测序技术鉴定了与热处理相关基因,Azim等[21]报道了芒果叶绿体基因组,这些研究丰富了芒果生物信息数据。有关芒果果实生长发育过程的转录组信息鲜有报道。本研究利用高通量测序技術平台illumina Hiseq 2 000对芒果发育成熟过程的果实进行转录组测序、拼接组装,再用生物信息学的方法对得到的Unigene进行注释和功能分类,以期为芒果功能基因的发掘利用、特异miRNA的鉴定及功能分析等奠定基础。
1 材料与方法
1.1 材料
试验于2012年3~7月在广东省雷州市覃斗镇芒果基地进行,试验材料为15年生的红芒6号芒果树,试验地土壤属砖红壤,株行距4 m×4 m,树体生长良好。分别于幼果期(花后50 d)、膨大期(花后80 d)、采收期(青熟)与成熟期(常温放置后成熟)采集芒果果实,每个时期采10~15个果,采集后迅速带回实验室,将果皮与果肉分开并用液氮速冻后保存于-80 ℃冰箱中备用。
1.2 方法
1.2.1 芒果果皮与果肉RNA提取 RNA提取参照Shan等[22]建立的方法。
1.2.2 芒果果实转录组测序 基于转录组学研究的优势,以红芒6号芒果为材料,取不同发育阶段的果皮与果肉样本,分别提取总RNA后等量混合用于测序,转录组测序委托深圳华大基因公司完成。提取的总RNA使用DNaseI消化DNA后,用带有Oligo(dT)的磁珠富集真核生物mRNA;加入打断试剂在Thermomixer中适温将mRNA打断成短片段,以打断后的mRNA为模板合成一链cDNA,然后配制二链合成反应体系合成二链cDNA,并使用试剂盒纯化回收、粘性末端修复、cDNA的3′末端加上碱基“A”并连接接头,然后进行片段大小选择,最后进行PCR扩增;构建好的文库用Agilent 2 100 Bioanalyzer和ABI StepOnePlus Real-Time PCR System质检合格后,使用Illumina HiSeqTM 2000进行测序。
1.2.3 数据组装及基因功能注释 将测序获得的原始数据去除接头序列、或N含量过高或低质量的序列,得到净读长,用Trinity软件按Min contig length 100,Group pairs distance 250,path reinforcement distance 85,Min kmer cov 2参数进行数据组装。将具有重叠区域的reads连成更长的Contig,采用Overlap的方法进一步拼接成Unigene。
通过Blastx程序将Unigene序列比对到蛋白数据库NR、Swiss-Prot、KEGG和COG(E值<0.00 001),并通过Blastn程序将Unigene与核酸数据库NT(E值<0.00 001)进行比对,获得Unigene最高序列相似性的蛋白,便得到该Unigene的蛋白功能注释信息。将Unigene与COG数据库进行比对,获得Unigene可能的功能注释及功能分类。根据nr注释信息,利用Blast2GO软件得到Unigene的GO注释信息,并对所有Unigene用WEGO软件进行GO功能分类统计,根据KEGG注释信息进一步分析得到Unigene的Pathway途径注释。
预测编码蛋白框:按NR、Swiss-Prot、KEGG和COG顺序将Unigene序列与以上数据库Blastx比对(E值<10-5),取Blast比对结果中排列最高的蛋白确定该Unigene的编码区序列,将编码区序列翻译成氨基酸序列,得到该基因编码区的核苷酸序列(序列方向5′→3′)和氨基酸序列。若Unigene比对不上以上数据库,用软件ESTScan[23]预测编码区,得到其编码区的核苷酸序列和氨基酸序列。
2 结果与分析
2.1 转录组组装结果
2.1.1 果实转录组组装概要 转录组测序共获得82 817 950读段,经过去除杂质和冗余处理,得到68 419 722条净读段,6.1Gb(6 157 744 980 bp)的有效数据,13×的测序深度,平均读长90 bp。其中读长大于20个碱基比例为95.27%,GC%值为45.52%,可以看出此次转录组测序结果较好,可为后续的数据组装提供很好的原始数据。转录组测序组装得到124 002个Contig,平均长度为338 bp,N50值为623 bp。进一步拼接得到54 207个Unigene,平均长度838 bp,N50值为1 328。按序列相似度对Unigene进行基因家族聚类,相似度大于70%的命为Clusters,共26 413个,Singleton序列有27 794个,其中E值小于10-5的序列共43 751个(表1)。54 207个Unigene的注释信息见http://www.sciencedirect.com/science/article/pii/S1874391914001523。
2.1.2 Contig和Unigene序列长度分布 组装的124 002个Contig中,核苷酸长度在100~200 bp的序列数有74 842,比例达到了60.36%;200~300 bp有17 520,占14.13%;300~400 bp有8 714,占7.03%;400~500 bp 的有4 538条,占3.66%;而≥ 500 nt的共有18 388条,比例为14.83%。由此可见,Contig片断长度以100~200 bp为主(表2)。在Contig数据的基础上,采用over-lap进一步拼接,共获得了54 207个unigene片段,其中核苷酸长度在100~500 bp的序列数有25 509,比例高达47.06%,500~1 000 bp有12 727,比例为23.48%;1 000~1 500 bp有7 312,占13.49%;1 500~2 000 bp有 4 334条,占8.01%;≥2 000 bp有4 315,占7.96%(表3)。
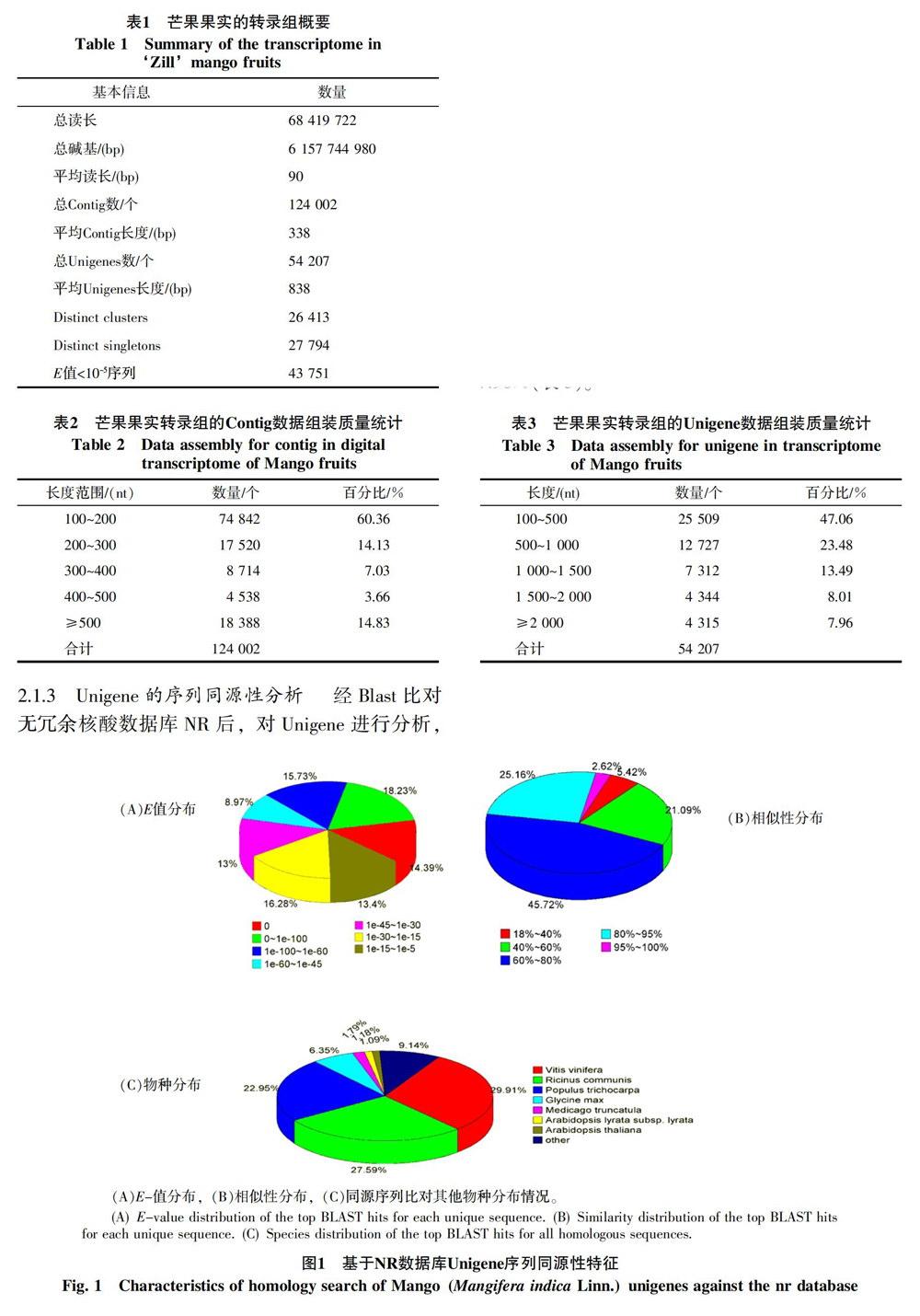
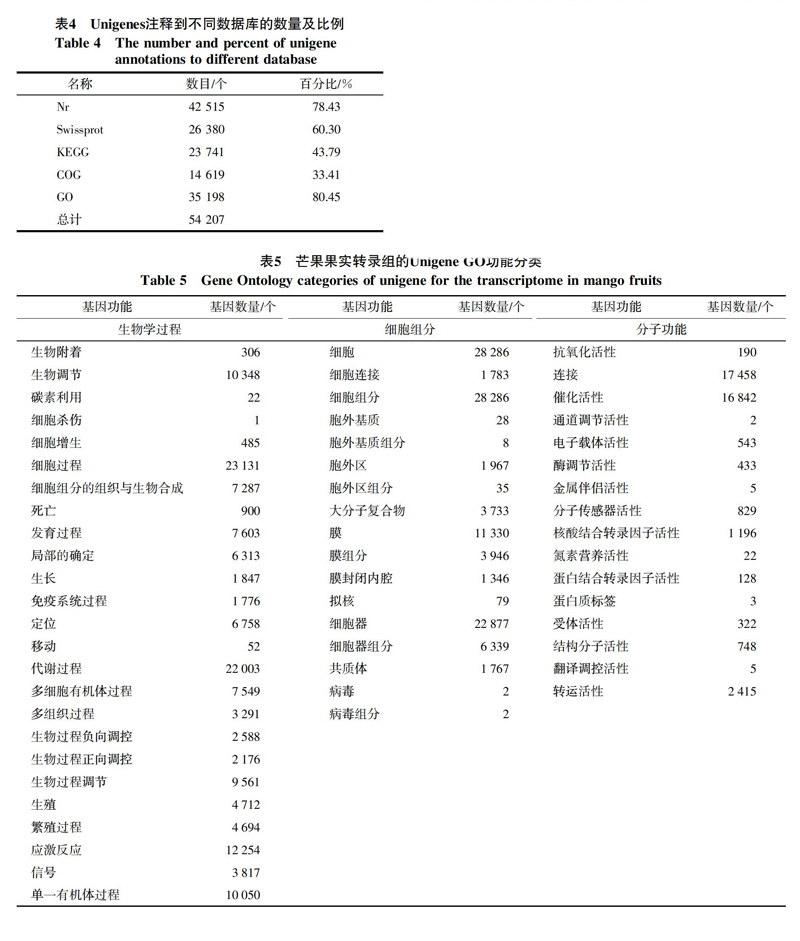
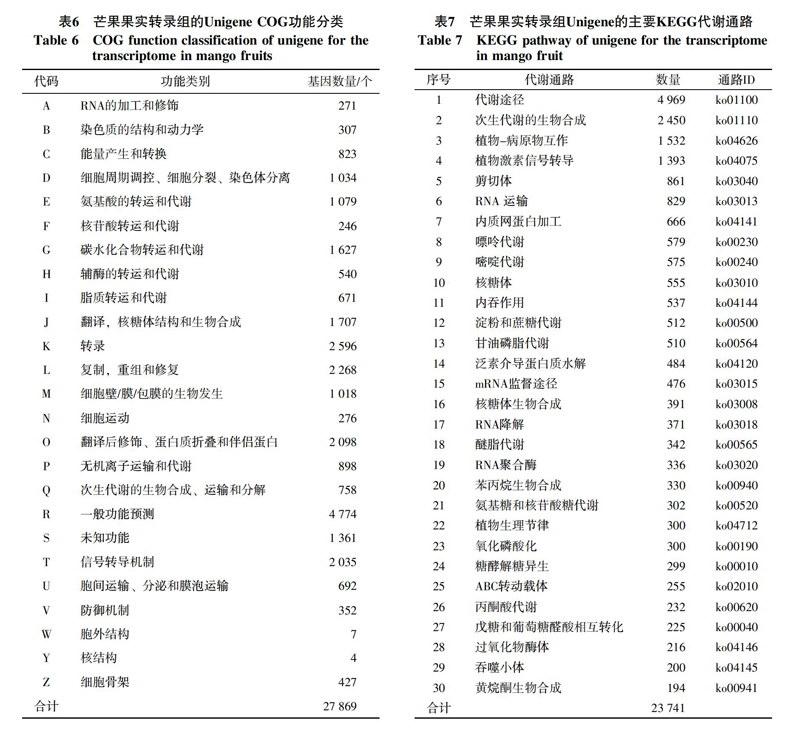
2.1.3 Unigene的序列同源性分析 经Blast比对无冗余核酸数据库NR后,对Unigene进行分析,从比对的E值来看(图1-A),发现29.68%的序列具有一定的同源性(10-30 序列的相似性比较结果表明,相似性高于80%的序列高达27.8%,相似性在60%~80%的序列高达45.7%,而26.5%的序列相似性在18%~60%间(图1-B)。对获得的 Unigene进行Blastn分析(图1-C),大部分Unigene序列和葡萄、蓖麻、毛果杨、大豆、苜蓿、拟南芥的同源序列比对上。统计显示29.9%的Unigene可比对到葡萄;27.6%的Unigene与蓖麻同源;依次为毛果杨(22.9%),大豆(6.3%),拟南芥(1.1%)等,这说明芒果与葡萄的进化关系较近。 2.2 Unigene功能注释 通过Blastx程序在不同的数据库进行搜索比对(表4),42 515条unigene比对到NR数据库,26 380条Unigene比对到Swissprot数据库,23 741条Unigene用于KEGG途径分析,14 619条进行COG分析,35 198条被用于GO分类。 2.2.1 Unigene的GO功能分类分析 12 923条Unigene与GO数据库中的基因具有相似性,且较多的单条Unigene与多种基因相对应,建立了302 479条对应关系,从而得到尽可能多的注释和分类。芒果果实转录组中的Unigene根据GO功能可分为生物学过程、细胞组分和分子功能3大类58分支(表5)。生物学过程类别中,细胞代谢过程和代谢过程涉及的基因最多,分别有23 131和22 003 条,细胞组分类别中,组成细胞和细胞部分涉及的基因最多,均为28 286条,分子功能类别中,结合功能涉及的基因最多,有17 458条,其次是具有催化活性功能的基因,达到了16 842条,其他种类基因的表达丰度不尽相同。 2.2.2 Unigene的COG功能分类 将Unigene和COG数据库进行比对,27 869条Unigene与数据库中的基因具有相似性,且较多的单条Unigene能够与多种基因相对应,建立了65 536条对应关系(表6)。Unigene根据功能大致可分为25类,Unigene的COG功能种类比较全面,涉及了大多数的生命活动,其中整体功能类的基因数量最多,有4 774 条,其次是转录功能(2 596),复制、重组和修复功能(2 268),翻译后加工、蛋白质折叠的伴侣蛋白(2 098),碳水化合物运输和代谢(1 627),核结构相关基因类的数量最少,只有4条,而未知功能的序列多达1 361条。COG分类结果表明这些Unigene涉及许多的生物学功能,这些未知基因可能参与芒果果实生长发育和成熟的调控。 2.2.3 Unigene的KEGG代谢途径分类 芒果果实转录组数据有23 741个Unigene能注释到128类代谢途径中(表7),包括代谢途径、次生代谢的生物合成通路、植物-病原互作、植物激素信号转导、DNA剪切、RNA运输、蛋白质在内质网的加工、淀粉和蔗糖代谢、苯丙氨酸的生物合成、萜类化合物合成,脂类代谢,RNA降解、黄酮和黄酮醇的生物合成等,其中代谢途径和次生代谢的生物合成所注释的Unigene数最多,分别为4 969和2 450。 2.3 编码蛋白框(CDS)的核苷酸和氨基酸序列分析
[5] Pandit, S S, Chidley, H G, Kulkarni, R S, et al. Cultivar relationships in mango based on fruit volatile profiles[J]. Food Chem, 2009, 114(1): 363-372.
[6] Pandit S S, Kulkarni R S, Chidley H G, et al. Changes in volatile composition during fruit development and ripening of ‘Alphonso mango[J]. J Sci Food Agr, 2009, 89(12): 2 071-2 081.
[7] Ma X W, Wu H X, Liu L Q, et al. Polyphenolic compounds and antioxidant properties in mango fruits[J]. SciHortic, 2011, 129(1): 102-107.
[8] MantheyJ A, Perkins-Veazie P. Influences of harvest date and location on the levels of beta-carotene, ascorbic acid, total phenols, the in vitro antioxidant capacity, and phenolic profiles of five commercial varieties of mango(Mangifera indica L.)[J]. J Agric Food Chem, 2009, 57(22): 10 825-10 830.
[9] Ornelas-Paz J D, Yahia E M, Gardea A A. Changes in external and internal color during postharvest ripening of ‘Manila and ‘Ataulfo mango fruit and relationship with carotenoid content determined by liquid chromatography-APcI(+)-time-of-flight mass spectrometry[J]. Postharvest Biol Tec, 2008, 50(2-3): 145-152.
[10] Jiang Y, Joyce D C. Effects of 1-methylcyclopropene alone and in combination with polyethylene bags on the postharvest life of mango fruit[J]. Ann Appl Biol, 2000, 137(3): 321-327.
[11] Schuster S C. Next-generation sequencing transforms today's biology[J]. Nat Methods, 2008, 5(1): 16-18.
[12] Shendure J, Ji H. Next-generation DNA sequencing[J]. Nat Biotechnol, 2008, 26(10): 1 135-1 145.
[13] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics[J]. Nat Rev Genet, 2009, 10(1): 57-63.
[14] Feng C, Chen M, Xu C J, et al. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq[J]. BMC Genomics, 2012, 13: 19.
[15] Vimolmangkang S, Zheng D, Han Y, et al. Transcriptome analysis of the exocarp of apple fruit identifies light-induced genes involved in red color pigmentation[J]. Gene, 2014, 534(1): 78-87.
[16] Liu G Q, Li W S, Zheng P H, et al. Transcriptomic analysis of ‘Suli pear (Pyrus pyrifolia white pear group) buds during the dormancy by RNA-Seq[J]. BMC Genomics, 2012, 13(1): 1-18.
[17] Sweetman C, Wong D C, Ford C M, et al. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression[J]. BMC Genomics, 2012, 13(1): 105.
[18] Yu K Q, Xu Q, Da X L, et al. Transcriptome changes during fruit development and ripening of sweet orange(Citrus sinensis)[J]. BMC Genomics, 2012, 13(1): 10.
[19] Dautt-Castro M, Ochoa-Leyva A, Contreras-Vergara C A, et al. Mango (Mangifera indica L.) cv. Kent fruit mesocarp de novo transcriptome assembly identifies gene families important for ripening[J]. Front plant sci, 2015, 6(10): 62.
[20] Luria N, Sela N, Yaari M, et al. De-novo assembly of mango fruit peel transcriptome reveals mechanisms of mango response to hot water treatment[J]. BMC Genomics, 2014, 15(1): 957.
[21] Azim M K, Khan I A, Zhang Y. Characterization of mango (Mangifera indica L.) transcriptome and chloroplast genome[J]. Plant mol biol, 2014, 85(1-2): 193-208.
[22] Shan L L, Li X, Wang P, et al. Characterization of cDNAs associated with lignification and their expression profiles in loquat fruit with different lignin accumulation[J]. Planta, 2008, 227(6): 1 243-1 254.
[23] Iseli C, Jongeneel C V, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences.[C]// Proc Int Conf Intell Syst Mol Biol, 1999: 138-148.
[24] Li X Y, Sun H Y, Pei J B, et al. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants[J]. Gene, 2012, 511(1): 54-61.
[25] Ong W D, Voo L Y, Kumar V S. De novo assembly, characterization and functional annotation of pineapple fruit transcriptome through massively parallel sequencing[J]. PLOS One, 2012, 7(10): e46937.
[26] Pandit S S, Kulkarni R S, Giri A P, et al. Expression profiling of various genes during the fruit development and ripening of mango[J]. Plant Physiol Biochem, 2010, 48(6): 426-433.
[27] Sherman A, Rubinstein M, Eshed R, et al. Mango (Mangifera indica L.) germplasm diversity based on single nucleotide polymorphisms derived from the transcriptome[J]. BMC Plant Biol, 2015, 15: 277.
[28] 羅 纯, 武红霞, 姚全胜, 等. 芒果转录组中SSR位点信息分析与引物筛选[J]. 热带作物学报, 2015, 36(7): 1 261-1 266.
[29] 王 明, 应东山, 王琴飞, 等. 基于454 GSFLX高通量测序的芒果微卫星标记特征分析[J]. 南方农业学报, 2015(10): 1 745-1 749.
[30] AltermannE, Klaenhammer T R. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes(KEGG) database[J]. BMC Genomics, 2005, 6: 60.
[31] Blencowe B J, Ahmad S, Lee L J. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes[J]. Genes Dev, 2009, 23(12): 1 379-1 386.
