高血糖通过抑制线粒体自噬加重大鼠脑缺血/再灌注损伤
2016-07-07左玮,梅丹
左 玮,梅 丹
(中国医学科学院北京协和医学院北京协和医院,北京 100730)
高血糖通过抑制线粒体自噬加重大鼠脑缺血/再灌注损伤
左玮,梅丹
(中国医学科学院北京协和医学院北京协和医院,北京100730)
摘要:目的探讨急性高血糖对局灶性脑缺血大鼠神经功能损伤的作用及机制。方法按照随机数字法,将80只♂ SD大鼠分为假手术组、脑缺血模型组(正常血糖NG)、脑缺血合并高血糖1组(HG1)、脑缺血合并高血糖2组(HG2)。线栓法建立大鼠局灶性大脑中动脉闭塞(MCAO)模型,缺血期间腹腔注射50%葡萄糖溶液诱导急性高血糖。MCAO 24 h后,采用Ludmila Belayev 12分法对大鼠的神经功能进行评分;采用TTC法对大鼠脑梗死体积和水肿程度进行评估;采用免疫荧光双标和电镜的方法对线粒体自噬的现象进行观察;进一步通过Western blot的方法对脑组织中自噬相关蛋白(LC3和Beclin-1),以及线粒体介导的凋亡相关因子(Cyt-C,AIF,caspase-9和caspase-3)的表达情况进行检测。结果正常血糖组、高血糖1组和高血糖2组的血糖分别控制在4、10和20 mmol·L-1的水平。与模型组相比,高血糖1组大鼠的神经功能评分和脑梗死体积差异没有显著性(P>0.05),而高血糖2组大鼠的神经功能损伤、梗死体积和水肿程度都有明显增加,差异具有统计学意义(P<0.05)。缺血/再灌注损伤3 d后取材进行检测,较模型组相比,高血糖2组大鼠的线粒体自噬水平减少,差异具有统计学意义(P<0.05),受损线粒体增加,伴随Cyt-C和AIF释放以及caspase-8/caspase-3活化的增加,差异具有统计学意义(P<0.05)。结论轻度急性期高血糖不加重脑梗死损伤。重度急性期高血糖则可能是通过抑制受损线粒体的自噬性清除,导致受损线粒体堆积,进而扩大线粒体介导的下游的凋亡损伤,加重了大鼠的脑梗死损伤程度。
关键词:脑梗死;高血糖;线粒体自噬;TTC染色;凋亡;神经功能;线粒体
脑血管疾病是危害人类健康的三大疾病之一,其中缺血性脑血管疾病占80%,并随着人口老龄化的增加,发病率呈明显上升的趋势。流行病学及临床的研究数据显示,50%左右的急性型缺血性脑卒中患者发病后伴随非糖尿病性的高血糖的现象,且高血糖可以加重缺血/再灌注后的脑损伤程度[1-2]。应用胰岛素纠正卒中伴随的高血糖状态是目前公认的最有前景的治疗方案。大量的临床和动物实验数据都证实,胰岛素对治疗卒中合并高血糖具有十分积极的作用,可以降低高血糖带来的危害,但作用机制仍不十分明确[3-4]。此外,血糖调控普遍存在的问题是,严格降糖治疗的效果仍缺乏循证医学的证据,胰岛素的治疗会伴随较大的低血糖风险,最佳的血糖水平目前还存在争议,卒中患者血糖实时监控的实施存在困难。由此可见,进一步深入了解高血糖加重缺血损伤的机制,寻找潜在的治疗靶点具有更为重要的意义。
因此,本实验通过线栓法建立大鼠大脑中动脉闭塞模型,腹腔注射不同剂量的50%葡萄糖诱导MCAO大鼠的急性高血糖状态(轻度高血糖和重度高血糖),模拟临床上非糖尿病患者的急性高血糖状态。观察不同血糖状态对MCAO大鼠的影响,并对其作用机制进行探讨,以期为临床卒中合并急性高血糖的治疗提供新的思路和依据。
1材料
1.1实验动物
SPF级健康♂ SD大鼠,体质量260~280 g。由北京维通利华实验动物技术有限公司提供(合格证编号:SCXK京2009-0007)。
1.2药品与试剂
葡萄糖和2,3,5-氯化三苯基四氮唑(TTC)(中国医药上海化学试剂公司)。Optium血糖仪和血糖试纸(雅培公司,美国)。山羊抗兔-LC3(1 ∶1 000)、山羊抗兔-Beclin-1(1 ∶1 000)和山羊抗小鼠-COX4(Sigma公司,美国)。山羊抗兔cyt-C(1 ∶1 000)、山羊抗兔AIF(1 ∶1 000)、山羊抗兔caspase-3和山羊抗兔capase-9(1 ∶1 000)(Santa cruz公司,美国)。辣根过氧化物酶偶联二抗和BCA蛋白测定试剂盒(中杉金桥生物技术有限公司,北京)。PVDF膜(Millipore公司,美国)。
2方法
2.1动物及分组
将动物按照随机数字法分为假手术组、模型组(N,4 mmol·L-1)、脑缺血合并高血糖1组(HG1,10 mmol·L-1)和脑缺血合并高血糖2组(HG2,20 mmol·L-1),每组20只。手术前动物禁食15 h,自由进水,室温保持25℃。
2.2MCAO模型及合并急性高血糖模型的建立
参照Longa法[5]略加改进,建立线栓法大鼠大脑中动脉闭塞模型(MCAO)。大鼠在5%恩氟烷、30%氧气/70%氮气下诱导麻醉,约5 min后将大鼠尾部提起无任何张力表示诱导成功。大鼠仰卧固定在手术台,在2%恩氟烷、30%氧气/70%氮气下维持麻醉。碘伏对颈部皮肤进行消毒后,于颈部正中作2 cm左右的切口,暴露气管和颈动脉三角区,用显微镊分离肌肉、筋膜、颈总动脉(common carotid artery, CCA)、颈内动脉(internal carotid artery, ICA)和颈外动脉(external carotid artery, ECA)。用动脉夹暂时夹闭CCA近心端和ICA,并在CCA远心端备线勿扎紧。电凝结扎ECA,剪断后将其拉下与ICA成一条直线。在距CCA分叉远端约2 mm处用眼科剪作细小斜向切口,将肝素浸泡过的线栓(直径0.26 mm)插入,收紧备线,松开ICA上的动脉夹,用镊子将栓线延ICA缓缓推进颅内大脑中动脉,有阻力时(CCA分叉处开始,插入深度约18 mm)停止(避免扎破血管造成蛛网膜下腔出血),提示线栓到达大脑中动脉起始处,阻断MCA供血相关区血流,将备线扎紧并记录栓塞开始时间。缝合肌肉组织和皮肤切口,栓线尾部留于体外。栓塞90 min后,缓慢退出线栓至CCA切口处,恢复MCA区血流供应。术中维持大鼠肛温37℃左右,直至动物苏醒。假手术组大鼠不插入栓线,其余操作与模型组相同。动物术毕清醒后进行行为学评分,未成模以及死亡大鼠不计入试验。合并急性高血糖组大鼠采用腹腔注射方法给予50%的葡萄糖溶液,模型组(正常血糖组)于术前5 min、缺血后45 min和90 min给予0.9%的生理盐水2 mL·kg-1。模型组合并高血糖1组和2组于术前5 min分别给予2.5 mL·kg-1和8 mL·kg-1的葡萄糖溶液;术后45 min和90 min均给予2 mL·kg-1的葡萄糖溶液。于第1次注射后以及术后的30 min和75 min尾尖采血并进行血糖测定,维持各组目标血糖值为4、10、20 mmol·L-1。
2.3神经功能评分
参照Ludmila Belayev 12分评分法对动物神经功能损伤情况进行评估[6-7]:① 提尾悬空(2分)0分:无明显神经功能缺失;1分:梗死对侧肢体轻度屈曲;2分:梗死对侧肢体屈曲明显;② 肢体放置,分为视觉亚试验(前方、侧方)和触觉亚试验(前方、侧方)(8分)0分:动物肢体放置反应正常;1分:反应延迟但不超过2s;2分:反应延迟且>2s;③ 本体觉亚试验(2分)0分:与对侧比肢体同样有力;1分:与对侧比肢体稍有力;2分:与对侧比肢体无力。
2.4梗死体积检测
缺血/再灌注(I/R)24 h后处死大鼠,迅速断头取脑并将脑组织置于0.1 mmol·L-1的PBS冰水混合液中进行冰浴。1 min后取出放于脑槽中,每间隔2 mm连续冠状方向切片,共切出6片。第1刀切在视交叉与脑前极的中点处,自第1刀起,向上连续切两片,向后连续切4片。最后迅速将脑片放置于1.5% TTC的PBS缓冲液里,避光孵育10 min,期间翻转脑片一次。显色后,正常脑组织为红色,梗死部位呈白色。将脑片移入4%多聚甲醛中固定24 h后拍照记录。应用Image J软件进行图像分析,计算梗死体积和水肿程度。梗死体积/%= [(梗死面积×2mm)/(2×健侧脑面积×2mm)]×100%。水肿程度/%=(患侧脑面积-健侧脑面积)/2×健侧脑面积×100%。
2.5免疫组织荧光双标染色
对脑组织进行冰冻切片,正常血清封闭后,分别加入兔抗-LC3和小鼠抗-COX4,4℃孵育过夜。次日弃去一抗,PBST洗3次后,分别加入二抗:Alexa 488-conjugated donkey anti-rabbit 和Alexa 594-conjugated donkey anti-mouse IgG(1 ∶300; Invitrogen),37℃避光孵育30 min。抗萃灭封片剂封片,激光共聚焦显微镜观察拍照,Image Pro Plus 6.0(IPP 6.0)进行图像分析。
2.6Western blot检测
于I/R 24 h处死动物,取缺血/再灌注侧皮层的脑组织称重,放入预冷的组织裂解液(1 ∶9),冰浴中进行匀浆。破碎后的组织匀浆于4℃ 12 000 r·min-1离心20 min,取上清。用BCA试剂盒对蛋白含量进行测定。每组取50 μg蛋白样品上样,SDS-PAGE凝胶电泳对样品进行分离。分离后进行转膜、封闭、最后加入一抗,4℃孵育过夜。d 2弃去一抗,TBST洗膜3次后加入二抗,室温摇床孵育2 h。把ECL混合液均匀加在膜上,LAS3000 image reader系统读取并拍照,保存图像,Quantity one 软件进行图片分析。
2.7透射电镜
各组动物分别于术后各时间点水合氯醛腹腔注射麻醉,立即断头取脑,分离右侧半球大脑皮质,大小约1 mm3,迅速置入预冷的2.5%戊二醛溶液中,4℃保存固定2 h后,0.1 mol·L-1的PBS洗3次。随后用1%锇酸后固定2 h。乙醇逐级脱水,树脂包埋。厚度为50 nm超薄切片。枸椽酸铅染色。HITACHI H-7650型透射电镜观察自噬体、溶酶体的形态及数量,并摄片。
2.8统计学处理
3结果
3.1MCAO造模急性期大鼠血糖水平的测定
如Tab 1所示,各组大鼠的基础血糖水平没有显著性的差异(P>0.05)。MCAO造模后不同时间点对血糖水平进行检测发现,正常血糖组动物血糖水平稳定维持在4 mmol·L-1左右。高血糖1组动物和2组动物的血糖水平则明显增加,分别稳定维持在10和20 mmol·L-1左右,组内比较差异不具有显著性(P>0.05),组间比较具有统计学意义(P<0.01)。

Tab 1 Levels of blood glucose at different±s,n=5)
**P<0.01vsN
3.2急性高血糖对MCAO大鼠神经功能的影响
I/R 24 h后,各组动物出现不同程度的神经功能障碍,如对侧前肢屈曲、左侧转圈等。利用Ludmila Belayev 12分评分法进行评估发现,模型组(正常血糖)、高血糖1组和2组的神经功能缺陷平分分别为4.8±0.99、5.2±1.17、9.6±1.05。高血糖2组动物神经功能受损程度高于正常血糖组和高血糖1组,且差异具有统计学意义(F=32.509,P<0.05);高血糖1组与模型组相比,对动物神经功能的损伤程度差异没有显著性(P>0.05,见Fig 1)。
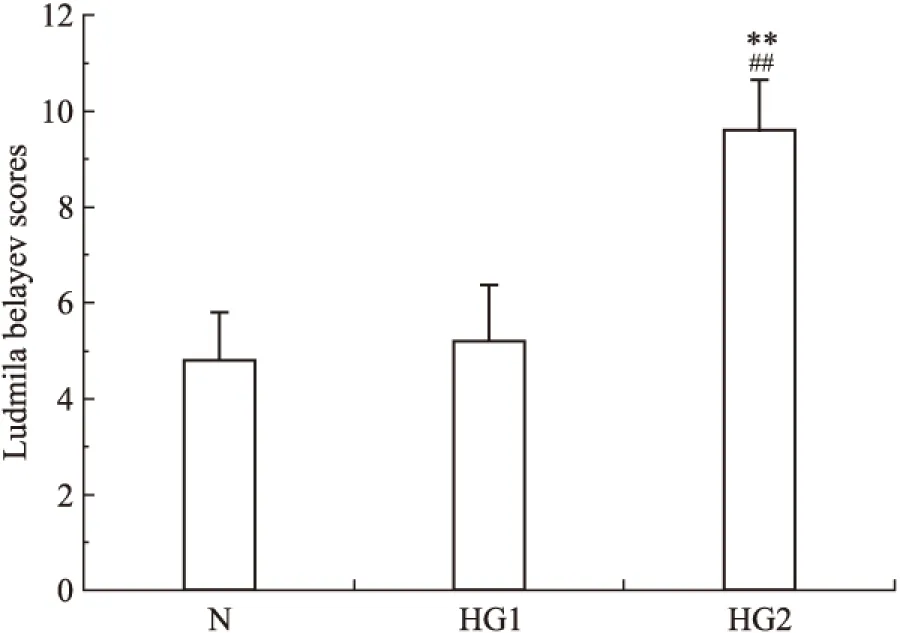
Fig 1 Effect of hyperglycemia on neurological
**P<0.01vsHG1;##P<0.01vsN.
3.3急性高血糖对MCAO模型大鼠脑梗死和水肿程度的影响
I/R 24 h后对脑组织进行TTC染色,评估脑梗死的情况。与假手术组(0%)相比,缺血损伤各组均有明显的梗死出现。模型组(N)、高血糖1组和2组的脑梗死体积百分比分别为28.6±0.99、29.2±1.17、33.2±1.05。与模型(N)组相比,高血糖2组(HG2)动物的脑梗死程度明显增加,差异具有统计学意义(F=58.9,P<0.05)。高血糖1组(HG1)组与模型组(N)组相比,脑梗死程度差异没有显著性(P>0.05)。(Fig 2A、B)。同样,与健侧脑组织相比,患侧脑组织出现明显水肿。模型组(N)、高血糖1组和2组的脑水肿体积百分比分别为14.8±0.89、15.1±1.47、19.6±1.07。与模型(N)组相比,高血糖2(HG2)组动物的脑水肿程度显著增加,差异具有统计学意义(F=52.7,P<0.01)。高血糖1组(HG1)与模型(N)组相比,脑水肿程度差异没有显著性(P>0.05)(Fig 2A、C)。
3.4急性高血糖对MCAO模型大鼠皮层线粒体自噬清除的影响
我们之前的研究发现,缺血后会短暂诱导受损线粒体的自噬性清除,是机体内源性的保护作用机制之一[8]。在此我们感兴趣的是高血糖是否对缺血诱导的线粒体自噬性清除有影响。我们通过荧光双标的方法,在I/R 24 h后对线粒体特异性标记物COX4和自噬小体的特异性标记物LC3Ⅱ进行共定位,观察线粒体自噬的情况。被LC3Ⅱ阳性信号包围的线粒体被认为是线粒体自噬[9]。从Fig 3中可以看到,较假手术组相比,模型组中被自噬小体包裹的线粒体数目明显增加(P<0.01)。高血糖1组与模型组相比,差异没有显著性(P>0.05)。而高血糖2组较模型组相比,则显著降低了线粒体自噬的水平(P<0.05)。我们进一步通过电镜的方法观察各组线粒体自噬水平的变化。从Fig 4中我们可以看到,正常的线粒体具有清晰完整的嵴结构和双层膜结构。与正常对照相比,模型组(N)在I/R 24 h后观察到大量的被双层膜自噬小体包裹的线粒体。而在高血糖2组中,镜下几乎观察不到线粒体自噬的现象,伴随高电子云密度和肿胀的受损线粒体[10]数目增加(P<0.01)。为了进一步确定高血糖诱导的缺血后线粒体自噬水平的降低是否是通过抑制大自噬流实现的,我们通过Western blot进一步对自噬相关蛋白LC3和p62的表达水平进行检测。从Fig 5实验结果中可以看到,模型组较假手术组相比,明显上调大自噬水平,差异具有显著性(P<0.01)。而高血糖组较模型组组相比,对大自噬流的影响差异没有显著性(P>0.05)。
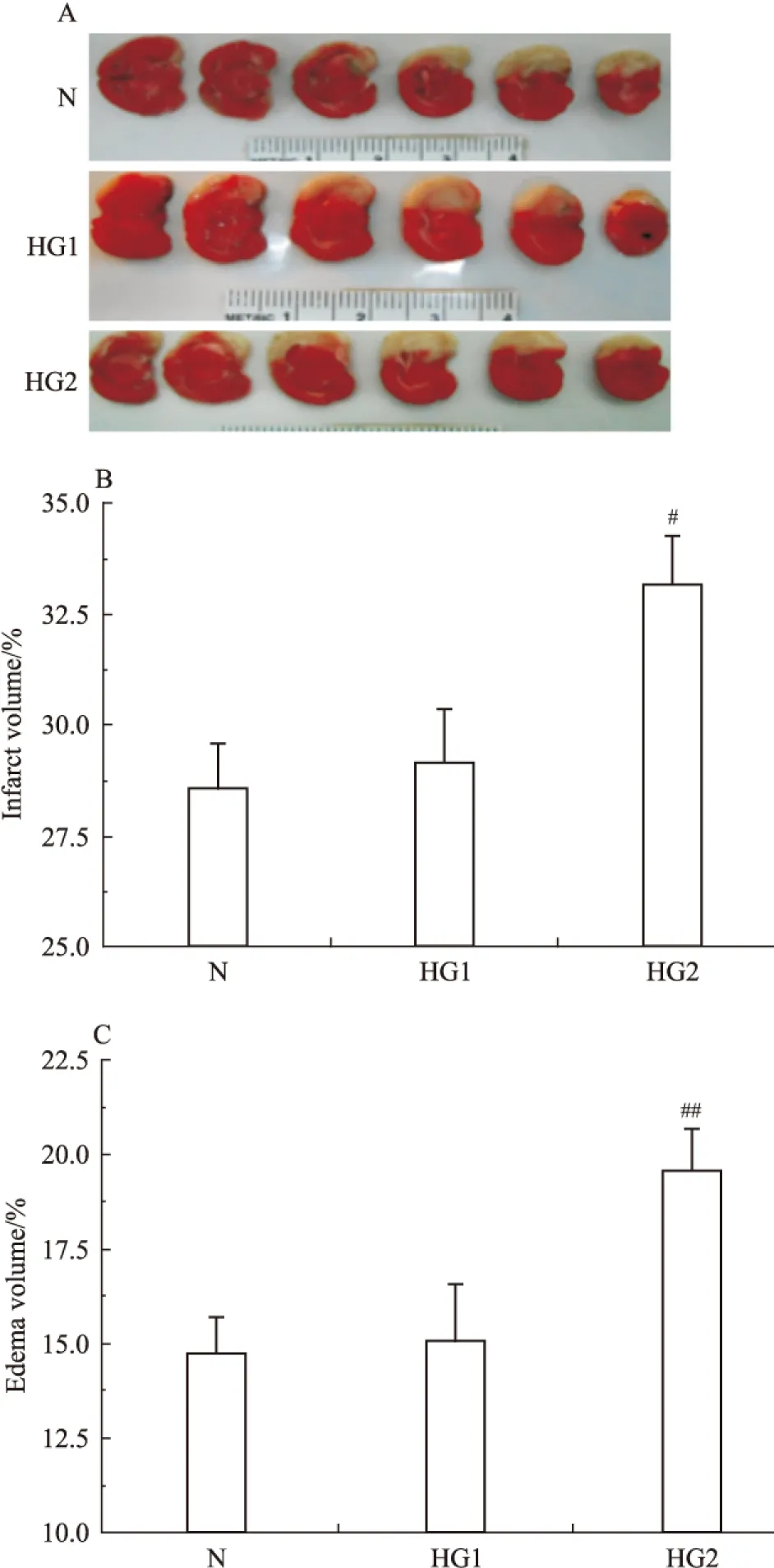
Fig 2 Effect of hyperglycemia on infarct volume
#P<0.05,##P<0.01vsN. A: TTC staining of brain sections; B: quantitative analysis of brain infarct volume;C: quantitative analysis of brain edema volume
3.5高血糖对MCAO模型大鼠脑内凋亡的影响
线粒体受损后会促进cyt-C的释放增加,促进凋亡的发生。从实验结果中可以看到,与模型组相比,高血糖1组中cyt-C的释放并没有增加(P>0.01),而高血糖2组cyt-C的释放明显增加,差异具有显著性(P<0.01)(Fig 6)。caspase-9和caspase-3是线粒体介导的凋亡信号通路的特异标记物,从实验结果中可以看到,与模型组和高血糖1组相比,高血糖2组caspase-9和caspase-3的活化明显增加,差异具有显著性(P<0.001)。

Fig 3 Effect of hyperglycemia on mitophagy in MCAO rats by immunoflurences±s,n=4)
##P<0.01vssham group;*P<0.05vsN. A: Representative double-staining images with COX4 and LC3 in ischemic penumbra;B: quantification of colocalization of LC3 and COX4.
4讨论
大量的临床研究和观察数据显示,急性血糖升高是脑卒中患者病情严重程度和预后不良的独立风险因素,缺血第1个24 h内血糖水平>7 mmol·L-1的患者与正常血糖患者相比,死亡率、致残率增高,神经功能预后更差[11]。另有研究发现,住院的缺血性卒中发病后血糖水平>6.1 mmol·L-1的非糖尿病患者,住院4周的死亡率是糖尿病患者的1.7倍[12]。美国糖尿病学会提出,包括缺血性脑卒中在内的所有住院高血糖非重症患者,都应积极控制空腹血糖小于7.8 mmol·L-1,而重症患者应通过胰岛素治疗,控制血糖在7.8~10 mmol·L-1之内[13]。但同时存在不同的观点认为,一定程度的血糖升高无需治疗,且是对卒中患者有益[14]。

Fig 4 Effect of hyperglycemia on mitophagy
##P<0.01vssham group;**P<0.01vsN group. Bar=500 nm.A:Representative EM images of mitochondrial autophagy in ischemic penumbra;B:Quantification of damaged mitochondria. Arrows indicate autophagosomes.
比如,在一项实验中,采用静脉输注葡萄糖-钾-胰岛素的方式对血糖进行严格的调控,发现降糖并不能减少患者的脑梗死体积[15],这也可能与入组患者血糖水平集中在7~12 mmol·L-1(轻度高血糖)有关。有关缺血性卒中后最佳的血糖水平目前仍无统一的定论,加之降糖所伴随的低血糖同时也是加重缺血性脑损伤的风险因素之一,因此为积极严格的血糖调控都带来困难。
动物实验数据显示,高血糖通过抗纤溶和促凝来影响溶栓疗效,破坏内皮功能,加重再灌注损伤。有研究发现,后适应可以对抗高血糖对缺血的损伤作用[16]但对于高血糖对缺血损伤的影响及作用机制尚未完全明确。我们前期的研究发现,缺血损伤可以短暂地诱导线粒体自噬的发生,这是机体内源性的保护作用机制之一,可以通过及时地清除受损线粒体,避免线粒体下游介导的凋亡级联损伤反应来发挥一定的保护作用[8]。高血糖是否对该过程具有一定的影响目前还不清楚。为了深入进行机制研究,本实验通过腹腔注射葡萄糖溶液诱导卒中发病后的高血糖状态,该方法可以很好的区别糖尿病性高血糖的状态,不涉及糖尿病伴随的复杂的病生机制,更好的模拟卒中后应激性的高血糖特征。从实验结果中可以看到,本研究中建立的轻度高血糖组(高血糖1组)的血糖水平能很好的模拟临床上轻度的血糖波动范围,其对MCAO大鼠的神经功能和梗死体积的影响与正常血糖组相比差异无显著性。而高血糖2组(重度高血糖)与模型组和高血糖1组相比,脑梗死体积和神经功能缺陷程度都有明显加重,并具有统计学意义。提示我们作为脑组织中重要的功能性物质之一,葡萄糖水平的轻度升高的可能不会加重急性缺血性脑损伤,但当其水平超出一定的限度,则会加重缺血缺氧性脑损伤。
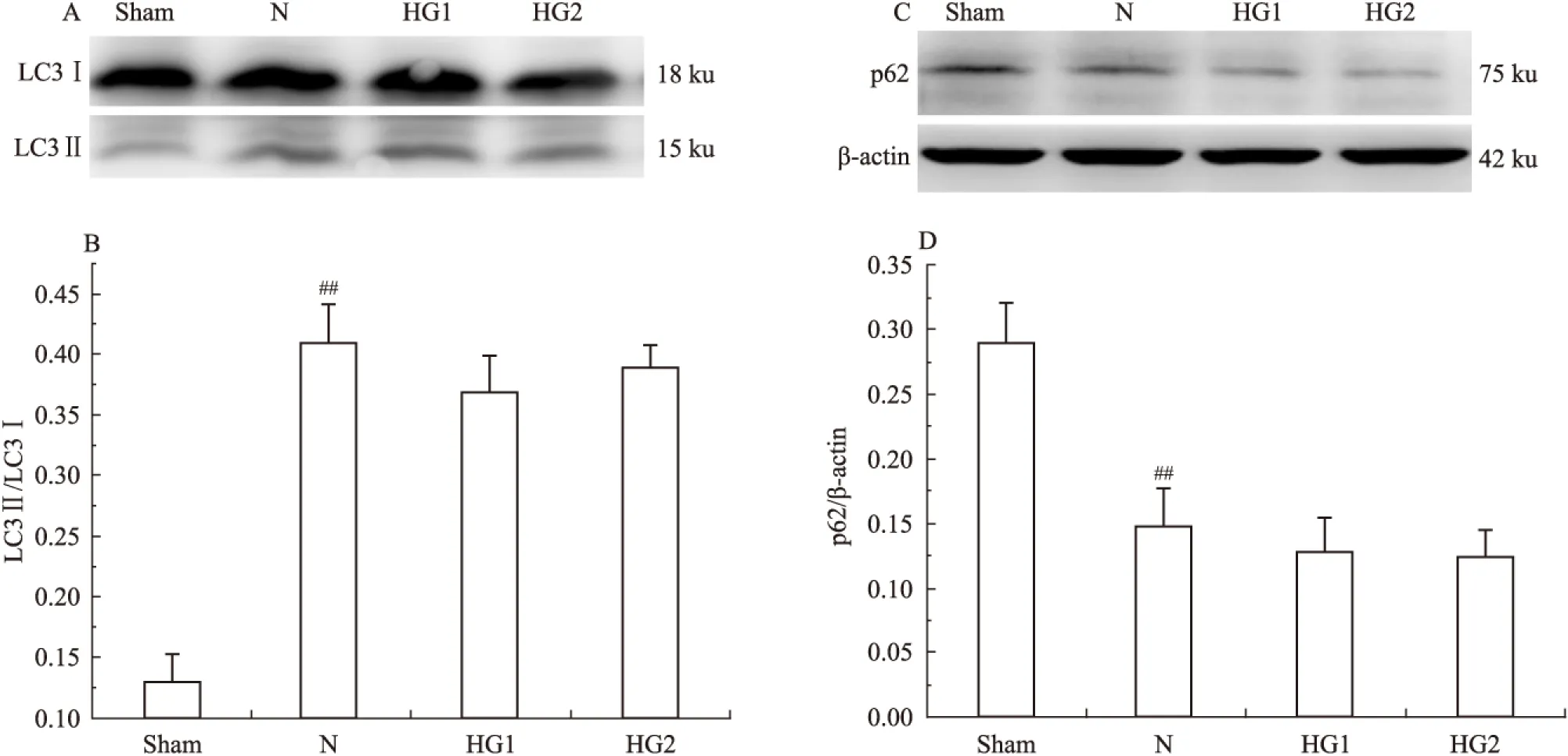
Fig 5 Effect of hyperglycemia on bulk autophagic flux in MCAO±s,n=3)
##P<0.01vssham group.AB:Western blot and quantitative analysis of LC3 expression;CD: Western blot and quantitative analysis of p62 expression.
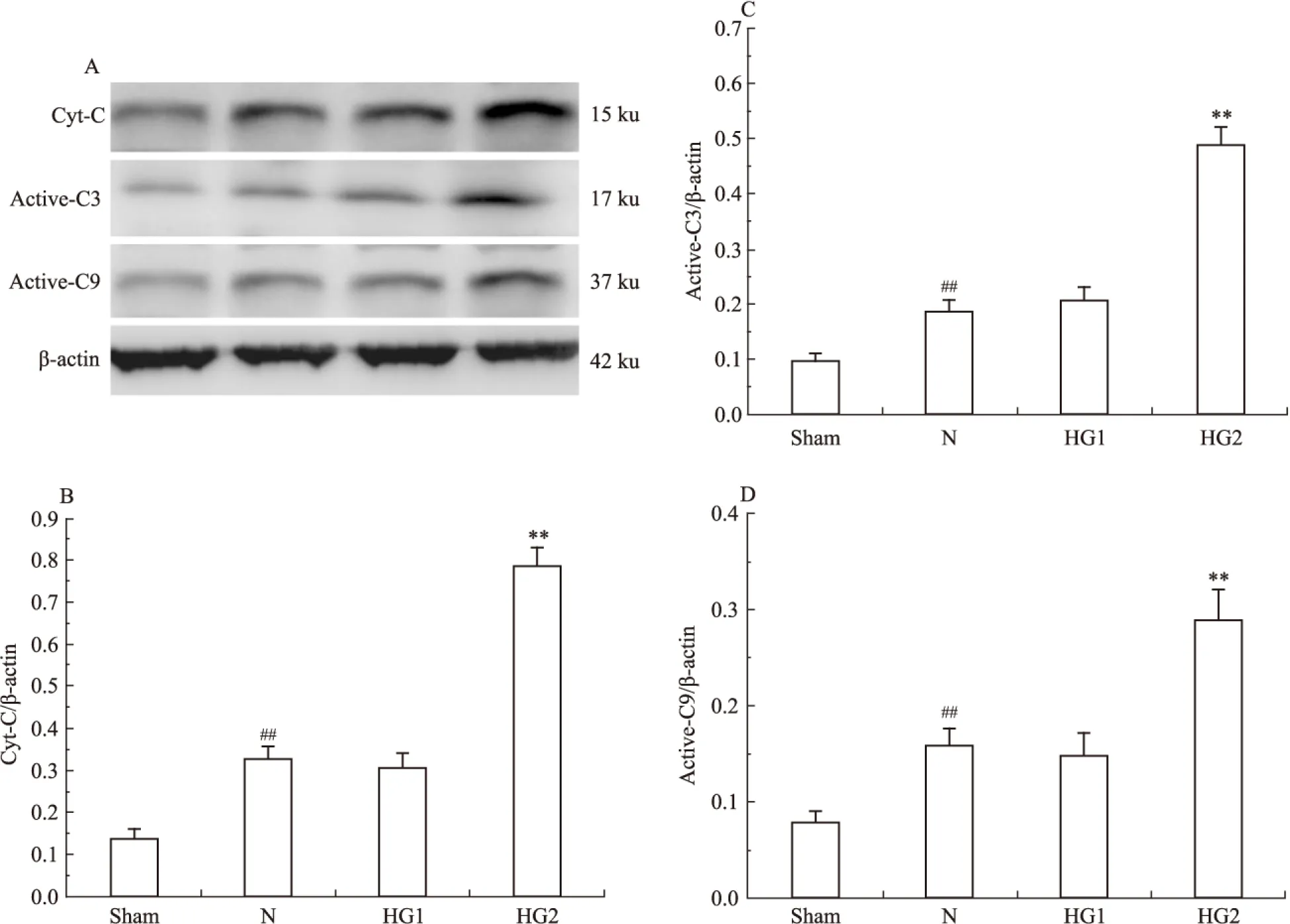
Fig 6 Effect of hyperglycemia on mitochondrial mediated apoptosis in MCAO±s,n=3)
##P<0.01vssham group;**P<0.01vsN group.A:Western blot of Cyt-C, Active-C3 and Active-C9.BCD: quantitative analysis of expression of Cyt-C, Active-C3 and Active-C9.
进一步对机制进行研究发现,重度高血糖抑制了急性缺血早期内源性的线粒体自噬清除,导致了大量的受损线粒体的堆积。线粒体受损如缺血缺氧损伤后,诱发下游一系列的级联反应[17-19],活化内源性凋亡,这也是造成缺血性中风后脑神经元损伤的重要原因之一。如促使线粒体释放细胞色素C(Cyt-C)的释放,Cyt-C在细胞质内与凋亡诱导因子1(AIF-1)、ATP,胱天蛋白酶原-9(pro-caspase-9)相互作用,形成凋亡小体,使得pro-caspase-9被激活为caspase-9,从而进一步激活胱天蛋白酶原-3(pro-caspase-3),使其被裂解为活性状态的caspase-3,引起细胞凋亡[19-20]。从我们的实验结果中可以看到,重度高血糖促进了Cyt-C的释放,扩大了线粒体介导的凋亡通路的激活。提示我们重度高血糖可能是通过抑制受损线粒体的自噬性清除,导致受损线粒体堆积,进一步扩大了受损线粒体下游的级联损伤反应,最终加重了缺血性脑损伤。
(致谢:感谢实验过程中中国医学科学院药物研究所药理实验室老师和同学的帮助。)
参考文献:
[1]Jia Q, Zheng X, Zhao X, et al. Abnormal glucose regulation in patients with acute stroke across China: prevalence and baseline patient characteristics[J].Stroke, 2012, 43(3): 650-7.
[2]Clark M E,Payton J E,Pittiglio L I. Acute ischemic stroke and hyperglycemia[J].CritCareNursQ,2014,37(2): 182-7.
[3]Ntaios G, Papavasileiou V, Bargiota A, et al. Intravenous insulin treatment in acute stroke: a systematic review and meta-analysis of randomized controlled trials[J].IntJStroke,2014,9(4): 489-93.
[4]Luitse M J, van Seeters T, Horsch A D, et al. Admiddion hyperglycaemia and cerebral perfusion deficits in acute ishchamic stroke[J].CerebrovascDis,2013,35(2): 163-7.
[5]Longa E Z, Weinstein P R, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats[J].Stroke,1989, 20(1): 84-91.
[6]Belayev L, Alonso O F, Busto R, et al. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model[J].Stroke,1996, 27(9):1616-22.
[7]刘家兰,邓礼娟,原欢欢,等.小鼠局灶性脑缺血模型行为学指标相关性研究[J].中国药理学通报,2012,28(3):430-5.
[7]Liu J L, Deng L J, Yuan H H,et al. Correlation between behavioral indicators in a mouse model of focal cerebral ischemia[J].ChinPharmacolBull, 2012, 28(3): 430-5.
[8]Geisler S, Holmström K M, Skujat D, et al. PINK1/Parkin mediated mitophagy is dependent on VDAC1 and p62/SQSTM1[J].NatCellBiol,2010,12(2):119-31.
[9]Kukat A, Kukat C, Brocher J, et al. Generation of rho cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses[J].NucleicAcidsRes,2008, 36(7): e44.
[10]Lanier W L. The prevention and treatment of cerebral ischemia[J].CanJAnaesth,1999, 46(5Pt2): R46-R56.
[11]Kes V B,Solter V V,Supanc V, Demarin V. Impact of hyperglycemia on ischemic stroke mortality in diabetic and non-diabetic patients[J].AnnSaudiMed,2007,27(5):352-5.
[12]American Diabetes Association. Standards of medical care in diabetes-2014[J].DiabetesCare,2014,37(Suppl 1): S14-S80.
[13]张晟奕,郭怡菁.卒中后高血糖机制及其管理策略[J].中华脑血管病杂志(电子版),2013,7(3):15-8.
[13]Zhang S Y, Guo Y J. The mechanism and control strategy of hyperglycemia in stroke patients[J].ClinJCerebrovasDis,2013,7(3):15-8.
[14]Mccormick M, Hadley D, Mclean J R,et al. Randomized, controlled trial of insulin for acute poststroke hyperglycemia[J].AnnNeurol,2010,67(5): 570-8.
[15]Chaitanya G V, Babu P P. Differential PARP cleavage:an indication of heterogeneous forms of cell death and involvement of multiple proteases in the infarct of focal cerebral ischemia in rat[J].CellMolNeurobiol,2009,29(4):563-73.
[16]陈刚领,徐岚溪,赵欢,等.肢体后适应对高糖状态下大鼠脑缺血/再灌注损伤的保护作用[J].中国药理学通报,2015,6(31):780-4.
[16]Chen G L, Xu L X, Zhao H,et al. Protective effects of limb remote ischemic postconditionting on ischemic stroke rats under hyperglycemia[J].ChinPharmacolBull, 2015, 6(31):780-4.
[17]Kroemer G, Zamzami N, Susin S A. Mitochondrial control of apoptosis[J].ImmunolToday, 1997, 18(1): 44-51.
[18]Loh K P, Huang S H, De Silva R, et al. Oxidative stress: apoptosis in neuronal injury[J].CurrAlzheimerRes,2006,3(4): 327-37.
[19]Sugawara T, Fujimura M, Noshita N, et al. Neuronal death/survival signaling pathways in cerebral ischemia[J].NeuroRx, 2004, 1(1):17-25.
[20]葛建彬,顾锦华,李梅,等.银杏内酯A对小鼠脑缺血/再灌注损伤的保护作用及其抑制NF-κB信号通路下调p53、caspase-3表达的机制[J].中国药理学通报,2012,28(8):1105-10.
[20]Ge J B, Gu J H, Li M, et al. Neuroprotective effects of Ginkgolide A on a mouse model of transient focal cerebral ischemia associated with inhibition of NF-κB signaling pathway and down-regulation of the levels of p53 and caspase-3[J].ChinPharmacolBull, 2012, 28(8): 1105-10.
Hyperglycemia aggravated cerebral ischemia/reperfusion injury by inhibiting mitophagy
ZUO Wei,MEI Dan
(PekingUnionMedicalCollegeHospital,ChineseAcademyofMedicalSciences&PekingUnionMedicalCollege,Beijing100032,China)
Abstract:AimTo investigate the role of hyperglycemia in cerebral ischemia/reperfusion(I/R) injury with a middle cerebral artery occlusion(MCAO) rat model and its mechanism.MethodsEighty healthy male SD rats were randomly assigned into sham group, I/R group (normoglycemia), hyperglycemic I/R groupⅠ(HG1) and hyperglycemic I/R groupⅡ(HG2). The cerebral I/R model was established by occluding the middle cerebral artery(MCA) in rats. Hyperglycemia was induced by intraperitoneal injection of 50% glucose solution. Neurological deficit was determined by Ludmila Belayev test; infarct size and brain edema were measured by TTC staining; mitophagy was observed by double immunofluorescent staining and electron microscope. The expressions of autophagy-related proteins(LC3 and Beclin-1)and apoptosis-related proteins(Cyt-C,AIF,caspase-9 and caspase-3)were examined by Western blot furtherly.ResultsBlood glucose level was controlled at 4 mmol·L-1(normoglycemia), 10 mmol·L-1(HG1) and 20 mmol·L-1(HG2) respectively. There were no significant differences between model group and HG1 group in neurological deficit scores, infarct volume and edema size(P>0.05). However, these indications in HG2 group were significantly increased compared with model group(P<0.05). After 3 days of reperfusion, the level of mitophagy was significantly reduced accompanied with increased mitochondria damages in HG2 group(P< 0.05),and the expressions of mitochondrial related apoptotic proteins(Cyt-C,AIF,caspase-9 and caspase-3) were significantly increased accordingly compared to model group.ConclusionsMild hyperglycemia can not intensify the cerebal ischemic injury. In contrast, severe hyperglycemia significantly aggravates the brain ischemic injury by inhibiting the removal of injured mitochondria in a manner of mitophagy, thus amplifying the mitochondrial mediated cascade damage responses.
Key words:brain ischemia; hyperglycemia;mitophagy;TTC staining;apoptosis; neurological function;mitochondrian
收稿日期:2016-02-28,修回日期:2016-03-30
作者简介:左玮(1986-),女,博士,助理药师,研究方向:神经药理学,E-mail:eileenzuo@163.com;
doi:10.3969/j.issn.1001-1978.2016.06.021
文献标志码:A
文章编号:1001-1978(2016)06-0846-08
中国图书分类号:R-332;R322.81;R329.24;R587.1;R743.31
网络出版时间:2016-5-25 15:39网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20160525.1539.042.html
梅丹(1962-),女,博士,主任医师,研究方向:医院药学,通讯作者,E-mail:meidanpumch@163.com
