Optimization and application of protein C-terminal labeling by carboxypeptidase Y
2016-07-04WenwenDuanYangZhangandGuoqiangXuJiangsuKeyLaboratoryofTranslationalResearchandTherapyforNeuroPsychoDiseasesJiangsuKeyLaboratoryofPreventiveandTranslationalMedicineforGeriatricDiseasesCollegeofPharmaceuticalSciencesSoochow
Wenwen Duan, Yang Zhang, and Guoqiang XuJiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases, Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, Jiangsu, China
Optimization and application of protein C-terminal labeling by carboxypeptidase Y
Wenwen Duan, Yang Zhang, and Guoqiang Xu
Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases, Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, Jiangsu, China
Abstract:Proteolytic cleavage is one of the post-translational modifications and plays important roles in many biological processes, such as apoptosis and tumor cell metastasis. The identification of the cleavage events can improve our understanding of their biological functions in these processes. Although proteomic approaches using N-terminal labeling have resulted in the discovery of many proteolytic cleavages, this strategy has its own inherent drawbacks. Labeling of protein C-termini is an alternative approach. Here, we optimized the labeling procedure in the profiling protein C-termini by enzymatic labeling (ProC-TEL) and improved the labeling efficiency for the positive isolation of protein C-terminal peptides and mass spectrometric identification. We applied this approach to a complex protein mixture from Escherichia coli and identified many C-terminal peptides and internal cleaved peptides from more than 120 proteins. From the identified cleavages, we found several previously known internal proteolytic cleavage sites and many novel ones which may play roles in regulating normal biological processes. This work provides a potential new way, complementary to the N-terminomics, for the identification of proteolytic cleavages in complex biological systems.
Keywords:carboxypeptidase Y, C-terminal labeling, ProC-TEL, proteolytic cleavage, proteomics
Introduction
Proteolytic cleavage is one of the most important post-translational modifications, which can alter protein localization[1], activate or deactivate enzymes[2], and induce apoptosis[3]. Many tumor cells have high expression level of proteases, which cleave protein substrates and regulate tumor cell proliferation, migration, metastasis, and invasion[4-6]. Identification of these cleavage events can elucidate their biological functions in these processes. It may also lead to the discovery of potential biomarkers for the early detection of cancer[7-8].
Once a protein is cleaved, neo-N or neo-C-termini are generated. Direct identification of these neo-termini from peptide mixture by shotgun proteomics is challenging due to the presence of a large number of non-terminal peptides. The strategy for the isolation and further identification of proteolytic cleavage sites is to label protein termini differentially from other parts. Several approaches targeting protein N-terminus for the proteomic identification of N-terminal peptides have been developed and utilized to determine proteolytic cleavages[9-17]. Although these approaches have their own advantages, such as simple chemical labeling of amino groups and high labeling efficiency, they also have inherent shortcomings[9,18]. For example, some peptides derived from protein N-termini are either too long or too short to be identified by mass spectrometry (MS)[11]. In many circumstances after a protein is cleaved, one or both of the fragments become short-lived[19], preventing them from further identification. Therefore, many proteolytic cleavages may not be identifiable by the N-terminomics.
Approaches targeting protein C-termini are alternatives for the identification of proteolyticcleavages. Most of these approaches used a negative selection strategy. This strategy removes non-C-terminal peptides and retains C-terminal peptides for further identification[17,20-21]. These approaches require relative high efficiency in the removal of non-C-terminal peptides. Another strategy is to directly label protein C-termini with an affinity tag for their positive isolation and proteomic identification[21]. The prototype of this approach, profiling protein C-termini by enzymatic labeling (ProC-TEL), was developed previously using commercially available reagents for the affinity labeling of protein C-termini[18]. In this approach, the transpeptidase activity of carboxypeptidase Y (CPY)[22-23]was utilized to label protein C-termini with a nucleophile, biocytinamide. The biotin was further used as a handle for the downstream affinity purification of the labeled C-terminal peptides. Here, we further improved the C-terminal labeling procedure and the isolation of the biotinylated C-terminal peptides in the ProC-TEL technique. Application of this improved approach in Escherichia coli (E. coli) cell lysate resulted in the identification of known and novel proteolytic cleavage sites which may play roles in regulating protein translation, oxidation, and reduction.
1 Materials and methods
1.1Protein sample preparation
E. coli, BL21 (DE3), was grown in 100 mL Luria Bertani (LB) media in a bacterial shaker at 37 °C overnight and the cell culture was centrifuged at 4 000×g and 4 °C for 10 min. The pellet was resuspended in 15 mL phosphate buffered saline (PBS) containing protease inhibitor cocktail (Roche) and sonicated with an ultrasonics processor on ice. The cell lysate was centrifuged at 15 000×g and 4 °C for 10 min and the supernatant was collected. BL21 lysate (about 1 mg/mL) was mixed with ten volumes of methyl esterification solvent (50 mmol/L HCl in methanol) and the mixture was incubated at room temperature for 24 h in a four-dimensional rotator. The organic solvent was removed by dialysis with a 3.5 kDa MWCO semi-permeable membrane (Spectra/Pro) against double-distilled water (ddH2O) with 10 mmol/L acetic acid and the BL21 protein sample was lyophilized for subsequent biotinylation.
For the preparation of myoglobin sample, myoglobin was dissolved in water at a concentration of 1 mg/mL, and then mixed with 10 volumes of methyl esterification solvent. The mixture was incubated at 25 °C for 24 h in a four-dimensional rotator and the organic solvent was removed by vacuum centrifugation.
Previous experiments have demonstrated that the best substrates for CPY-catalyzed transpeptidation are proteins or peptides containing a carboxyl ester at their C-termini[18]. If a protein has a free carboxyl group at the C-terminus, the labeling efficiency is essentially undetectable. Therefore, methyl esterification was performed prior to the labeling.
1.2C-terminal labeling of the methyl esterified BL21 lysate
The CPY (Sigma)-catalyzed biotinylation was carried out according to a method previously described[18]with the following modifications. Sodium borate (Na2B4O7) buffer (0.1 mol/L) with indicated concentration of sodium dodecyl sulfate (SDS) was used for the biotinylation of methylated and lyophilized BL21 lysate. The reaction was carried out at pH 11.5. In order to study the effect of SDS on the overall efficiency of CPY-catalyzed biotinylation, the methyl esterified BL21 lysate was dissolved in 50 mmol/L sodium acetate (pH 5.5 and without SDS) and centrifuged at 14 000×g for 15 min to obtain the BL21 lysate. The reaction buffer contains 0, 0.1%, 0.2%, 0.5%, 1.0% or 1.5% (W/V) SDS. The labeling was carried out with the same volume of reaction buffer and protein sample in the presenceof 8 mmol/L biocytinamide (Bachem Biotechnology) and 1 μg/mL CPY at 37 °C for 1 h. The final SDS concentration in the reaction is about half of that in the initial reaction buffer. The biotinylated samples were blotted with Strep-HRP (Beyotime) for the detection of biotinylated proteins with immobilon Western chemiluminescence HRP substrate (Millipore).
In order to investigate the effect of SDS concentration on the solubility of methylated BL21 lysate, we used sodium acetate (50 mmol/L, pH 5.5) buffers containing different SDS concentration to dissolve the methyl esterified proteins after lyophilization. We ran SDS-PAGE and used silver staining to detect the overall solubility of the methylated proteins. The results from these experiments were used to determine the optimal concentration of SDS for the CPY-catalyzed labeling of protein C-termini. The subsequent experiments of BL21 lysate were conducted with the optimized labeling condition.
1.3C-terminal labeling of the methyl esterified myoglobin
In order to test whether the improved C-terminal labeling technique can be used to isolate C-terminal peptide from a single protein, we used myoglobin as a model protein. The methyl esterified myoglobin was mixed with the same volume of reaction buffer in the presence of 8 mmol/L biocytinamide and 1 μg/mL CPY. The reaction was carried out in a 0.1 mol/L Na2B4O7buffer (pH 11.5) at 37 °C for 1 h. The biotinylated samples were blotted with Strep-HRP to evaluate the labeling efficiency.
1.4SDS-PAGE and in-gel trypsin digestion
The biotinylated myoglobin and BL21 lysate were mixed with 1/4 volume of 5×SDS loading buffer containing 2.5% β-mercaptoethanol and heated at 100 °C for 10 min. A small fraction of biotinylated samples were used to validate the biotinylation efficiency by Western blotting with Strep-HRP. The rest of the samples (about 200 μg) were digested with sequencing grade modified trypsin (Promega) according to an in-gel trypsin digestion protocol after disulfide reduction and thiol alkylation[24-25]. The reduction was carried out with 10 mmol/L dithiothreitol at 50 °C for 30 min and the alkylation was performed with 50 mmol/L chloroacetamide at 25 °C for 45 min. Both reactions were performed in a buffer containing 25 mmol/L ammonium bicarbonate. Under this reaction condition, disulfide bonds were reduced and the free thiols were blocked. Therefore, they will not affect the downstream identification of C-terminal peptides by mass spectrometry. The resulting peptide mixture was then extracted from gel and dried in a vacuum centrifuge.
1.5Sample preparation for mass spectrometry analysis
In order to reduce the complexity of the samples before the subsequent MS identification, the neutravidin agarose beads (Thermo Scientific) were used to isolate the biotinylated C-terminal peptides. The dried peptide samples from myoglobin and BL21 lysate were dissolved in a 0.6 mL of binding buffer (3 mol/L urea and 1 mol/L NaCl in PBS) and incubated with 20 μL of neutravidin agarose beads in a 1.5 mL eppendorf tube for 3 h at room temperature. The beads were washed with 1.0 mL of following washing buffers (each with a 7 min incubation): 2×WB1 (8 mol/L urea in PBS), 2×WB2 (6 mol/L GdnHCl in PBS), 2×WB3 (6 mol/L urea and 1 mol/L NaCl in PBS), 2×WB4 (4 mol/L urea and 1 mol/L NaCl in PBS), 1×WB5 (10% isopropanol and 10% ethanol in 50 mmol/L ammonium bicarbonate), 1×WB6 (20% methanol in 50 mmol/L ammonium bicarbonate), ddH2O. The peptides were eluted by incubating the beads in 50 μL elution buffer (50% acetonitrile and 0.1% trifluoroacetic acid, TFA) for 10 min with constant shaking. The elution step was repeated once and the samples were combined, dried in a vacuum centrifuge, and desalted with a C18 ziptip (Agilent).
1.6Mass spectrometry analyses of peptides derived from myoglobin and BL21 lysate
The peptide samples from myoglobin were dissolved in a 50% acetonitrile solvent containing 0.1% TFA and α-cyano-4 hydroxycinnamic acid (Fluka). The relative molecular weights were measured in a matrix-assisted laser desorption/ionization and time of flight mass spectrometer (MALDI-TOF MS, Bruker). The purified peptides from myoglobin and the peptide mixtures from BL21 were analyzed on an Orbitrap Elite hybrid mass spectrometer (Thermo Scientific) coupled with a Dionex liquid chromatography. The solvent A is 0.1% formic acid in ddH2O and solvent B is 80% acetonitrile and 0.1% formic acid. The samples were loaded to an enrichment column (Acclaim PepMap100, C18, 5 µm, 100 Å, 300 µm i.d. ×5 mm) with a 2% acetonitrile and 0.05% formic acid and separated in an analytical column (Acclaim PepMap100, C18, 3 μm, 100 Å, 75 μm i.d. ×15 cm, nanoViper). The liquid chromatography gradient for peptide analysis is: 0−10 min, 6% solvent B; 10.1−130 min, 6%−44% solvent B; 130.1−140 min, 98% solvent B; 140.1−150 min, 6% solvent B. Mass spectra were acquired in the positive-ion mode with automated data-dependent MS/MS in the collision-induced dissociation mode for the 15 most intense ions from each precursor MS scan. Each selected precursor ion was analyzed twice within 60 s. The resolution for the precursor ion was set for 120 000 and the MS/MS spectra were collected within a 2 Da mass isolation window of the selected precursor ions.
1.7Data analysis
The MS/MS data were searched against the E. coli (strain K12) protein database from the UniProt database[26](released on October 22, 2014) using Proteome Discoverer 1.4 (Thermo Scientific) with a mass tolerance of 10 ppm for precursor ions and 0.6 Da for fragment ions. Cysteine carboxyiodomethylation was set as the fixed modification while methionine oxidation and C-terminal biocytinamide modification were set as variable modifications. Semi-trypsin cleavage was selected and the maximal number of trypsin missed-cleavages was set as two. Peptides within 1% false discovery rate (FDR) were exported to Excel sheet. The peptides between 1% and 5% FDR were only included if the spectra contain fragments from biocytinamide and matched the theoretical spectra very well. Only peptides with charge states of 2, 3, and 4 were included. All the MS/MS spectra were manually validated and the spectra with low quality fragmentations or low signal to noise ratio were discarded. Peptides with non-K/R residue proceeding to the N-termini were removed to make sure that the peptides were derived after tryptic cleavage at their N-termini.
1.8Bioinformatic analyses
We used the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources[27]to explore the cell compartment distribution for proteins identified with internal cleavages in our list. The UniProt accession numbers for these proteins were uploaded to the DAVID database and the cell compartment and biological processes were selected for the analyses. The statistically significantly enriched cell compartments were plotted against log(1/p-value). Other statistic analyses were carried out by counting the identified peptides or using the protein sequences extracted from the UniProt database for proteins of interest.
2 Results and discussions
2.1Determination of the optimal SDS concentration for the CPY-catalyzed biotinylation

Fig. 1 Optimization of protein C-terminal labeling by carboxypeptidase Y (CPY). (A) The effect of SDS concentration in the final reaction condition on the overall efficiency of CPY-catalyzed biotinylation. (B) Silver staining of carboxyl methylated proteins from E. coli dissolved in buffers containing different SDS concentration. Buffer composition: 50 mmol/L sodium acetate (pH 5.5) with indicated SDS concentration. (C, D) Effect of SDS in the final reaction condition on the efficiency of CPY-catalyzed C-terminal biotinylation. Samples from E. coli were carboxyl methylated, labeled with biocytinamide, blotted with Strep-HRP, and visualized with immobilon Western chemiluminescence HRP substrate. (C) is the short exposure and (D) is the long exposure.
In the original ProC-TEL approach, 0.1% SDS was included in the reaction buffer for C-terminal labeling[18]. However, it was found that the fraction of biotinylated proteins was relative low. CPY is a relative stable protease[28], which can function under relative harsh conditions, such as at relative low or high pH, in the presence of low concentration of detergents or denaturants. First, we examined the effect of a denaturant, urea, on the CPY-catalyzed biotinylation. Different concentrations of urea (0, 1, and 2 mol/L) were used in the biotinylation reaction, which was carried out at pH 11.5 and 37 °C for 1 h. The samples were blotted with Strep-HRP for the detection of biotinylated proteins from BL21 lysate. The Western blotting result showed that the overall efficiency of biotinylation decreases with the increase of the urea concentration (See supplementary Fig. S1 online). Then, we tested the effect of SDS concentration on theoverall efficiency of CPY-catalyzed biotinylation by using different SDS concentration in the reaction buffer but no SDS in the protein sample. Western blotting analysis showed that the biotinylation efficiency increases when the SDS concentration changes from 0 to 0.5% and decreases when SDS reaches 0.75% (Fig. 1A). This result implies that we may increase the SDS concentration up to 0.5% to increase the solubility of the methylated proteins and to subsequently improve the overall labeling efficiency. We then used buffers containing different SDS concentration to dissolve the methyl esterified proteins after lyophilization. Silver staining showed that upon the increase of the SDS concentration up to 1%, more methyl esterified proteins were dissolved (Fig. 1B). Our biotinylation experiments for these samples showed that 0.25% and 0.5% SDS in the final reaction condition can significantly increase the overall biotinylation efficiency (Fig. 1C and 1D for a long exposure). The 0.5% SDS reaches the balance between protein solubilization and CPY denaturation and is the optimal condition found for the C-terminal labeling. Therefore in the subsequent experiments, buffers containing 1% SDS were used to dissolve the esterified proteins and 0.5% SDS in the final reaction condition instead of 0.1% SDS in the reaction buffer in the original procedure[18]was used in the C-terminal labeling.
2.2Isolation of the C-terminal peptide from a protein through CPY labeling
We asked whether this procedure can be applied to isolate the protein C-terminal peptide from a protein so that the complexity of a sample can be dramatically reduced in the subsequent MS identification. The overall procedure is depicted in Fig. 2A. We first methyl esterified a model protein and performed CPY-catalyzed biotinylation, then digested it with trypsin, and further used neutravidin to purify the biotinylated peptides for MS analysis. We chose myoglobin to test this procedure. Consistent with a previous experiment[18], methylated myoglobin can be readily modified by biocytinamide in the presence of CPY, which was visualized by Western blotting of the conjugated biotin using Strep-HRP (Fig. 2B). The MALDI-TOF MS detected several peaks from myoglobin after trypsinolysis (the top panel in Fig. 2C). The relative molecular weights of two peaks (with solid and open circles), which are in the relative low abundance, match the C-terminal peptides of myoglobin with a biocytinamide. One of them is the completely cleaved tryptic peptide while the other contains one trypsin missed-cleavage site. After neutravidin purification, only two major peaks from biotinylated peptides were detected. In addition, the tandem MS of these two peaks demonstrated that they were indeed the C-terminal peptides from myoglobin (Fig. 2D). The MS/MS contained two signature peaks derived from the fragmentation of biocytinamide (labeled with BF and y0), which can be used for further validation of the MS/MS spectra[18]. These results demonstrated that the improved ProC-TEL procedure along with affinity purification and MS analysis can efficiently identify protein C-terminal peptides. It is consistent with our previous experiment for bovine pancreatic ribonuclease A[18]except that an additional peptide with a trypsin missed-cleavage site was identified for myoglobin.
2.3Identification of protein C-terminal peptides from a complex protein mixture
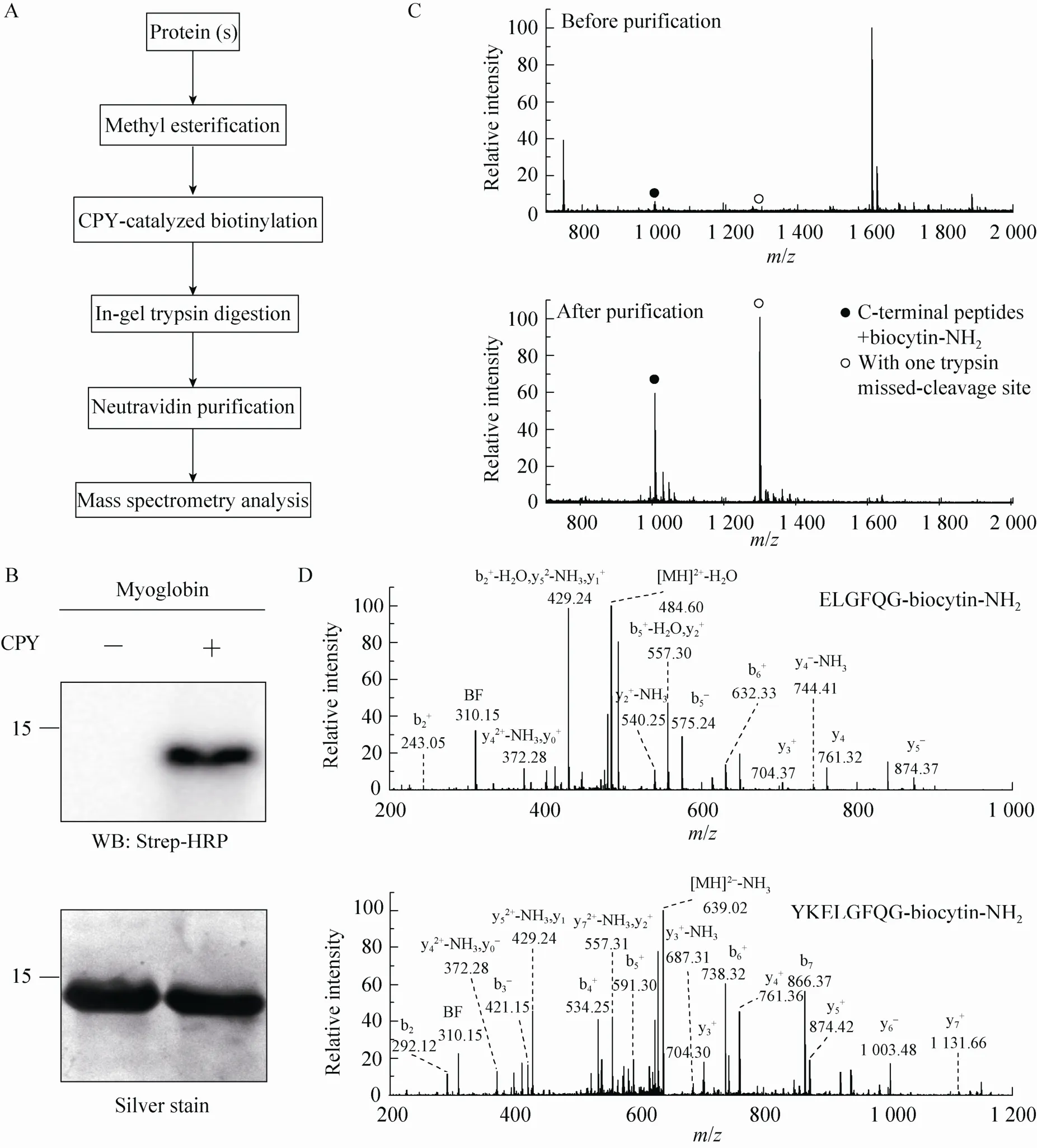
Fig. 2 Isolation of C-terminal peptides from myoglobin through modified ProC-TEL technique and neutravidin purification. (A) Flowchart of CPY catalyzed protein C-terminal labeling and sample preparation for MS analysis. (B) Western blotting and silver staining of carboxyl methylated myoglobin after CPY-catalyzed biotinylation. (C) MALDI-TOF MS of tryptic peptides (top panel) and affinity purified C-terminal peptides (bottom panel) obtained from biotinylated myoglobin. The biotinylated peptides derived from protein C-terminus are denoted by solid and open circles. The open circle indicates the peptide with one trypsin missed-cleavage site. (D) MS/MS spectra of affinity purified C-terminal peptides detected in the bottom panel of (C). The top and bottom panels are the MS/MS of C-terminal peptides without and with one trypsin missed-cleavage site, respectively. Peptide sequences are shown at the upright corner of the spectra.
A protein mixture from E. coli was used for this experiment. The SDS-PAGE gel containing BL21 cell lysate after biotinylation was excised to several gel pieces and the subsequent experiments were carried out in parallel for each gel piece (Fig. 3A). Through this experiment, we identified 122 peptides whose C-termini were modified by biocytinamide (See Supplementary Table S1 online). About 38% of the detected peptides are C-terminal biotinylated peptides. The majority of these peptides (91, 75%) are derived from C-termini of intact proteins. The MS/MS spectrum of a typical C-terminal peptide(top panel of Fig. 3B) contains two fragments from biocytinamide. In our identified C-terminal peptides, more than 97% have such fragments, which further validate their correct identification. In addition, we also identified many C-terminal peptides (31, 25%) derived from protein internal cleavages (Table 1 and bottom panel of Fig. 3C for a typical MS/MS spectrum). A few peptides are from previously known protein cleavages. For example, the C-terminal biotinylated peptide, VTHADLHYEG, is from aspartate 1-decarboxylase after the cleavage of its proprotein at the 24th amino acid. However, many biotinylated peptides are derived from internal cleavages currently unknown based on the annotation in the UniProt protein database. This result demonstrated that the modified ProC-TEL procedure can potentially be used to identify proteolytic cleavages.
In the preparation of BL21 cell lysate, protease inhibitor cocktail was used to inhibit protease activity. The BL21 lysate was further methyl esterified with methanol in the presence of 50 mmol/L HCl. Under such conditions, the endogenous proteases would be deactivated. In addition, the CPY catalyzed labeling of protein C-terminus was carried out at pH 11.5 and most endogenous proteases would be completely denatured at such a high pH. Therefore, under our experimental condition, the identified internal cleavages would most probably reflect the proteolytic cleavages occurred in cells.
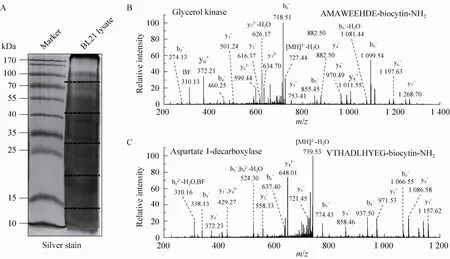
Fig. 3 ProC-TEL identifies protein internal cleavages from E. coli. (A) Typical silver staining of the biotinylated sample from E. coli. The gel was excised to several pieces for subsequent experiments. (B, C) MS/MS spectra of a typical C-terminal biotinylated peptide (B) and a peptide from an internal cleavage (C). The internal biotinylated peptide in (C) is derived from the C-terminus of the α subunit generated after the proprotein of aspartate 1-decarboxylase is cleaved at the carboxyl side of the 24th amino acid.
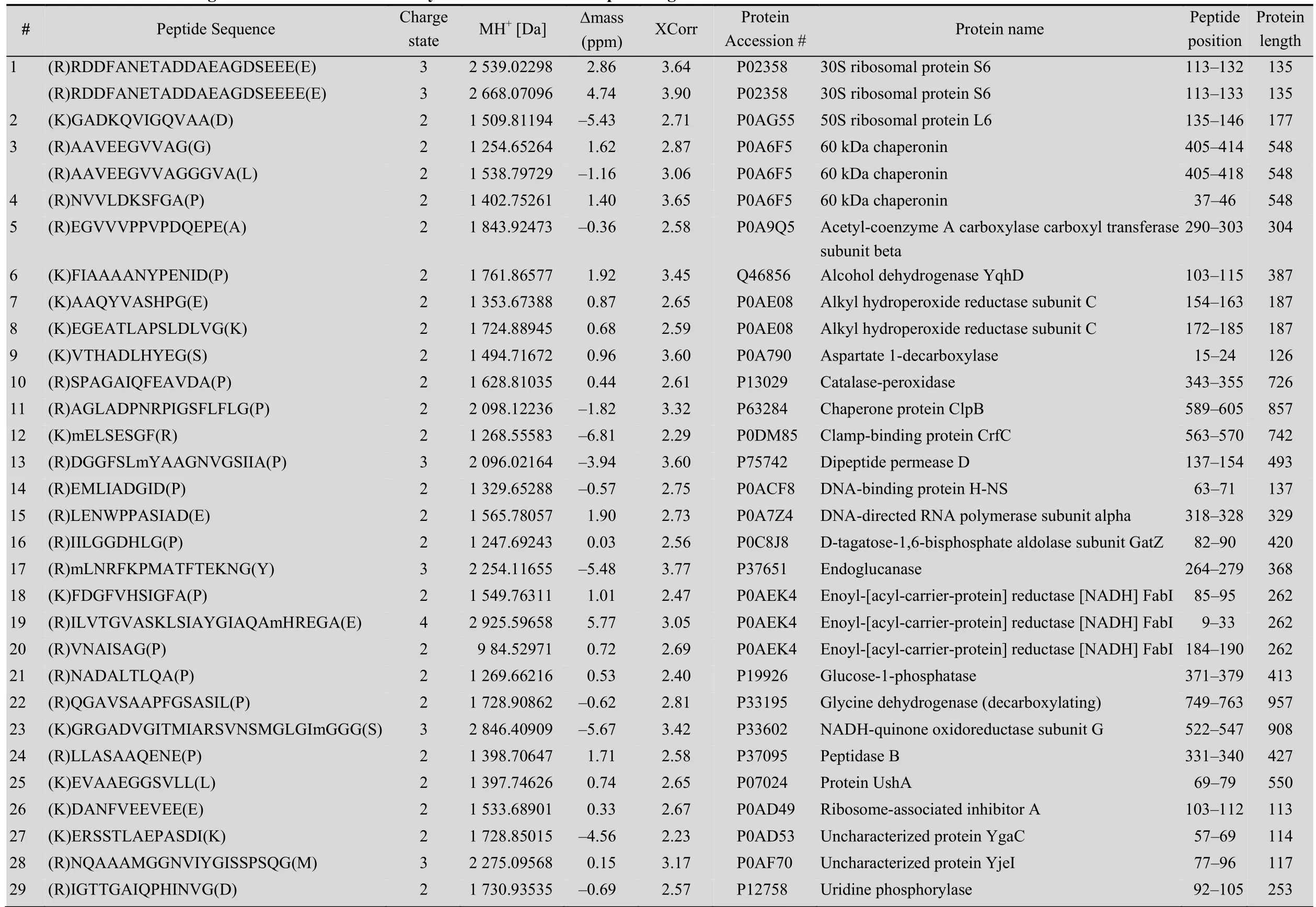
Table 1 Internal cleavages from E. coli identified bym odified ProC-TEL profiling
2.4Bioinformatic analyses of internal cleavages identified from E. coli lysate
In our experiments, we have identified 31 non-C-terminal peptides from E. coli (Fig. 4A), which are derived from the internal cleavages. Examination of these peptides showed that the majority of them are derived from internal cleavages far away from protein C-termini. Only a small number of these peptides are from C-terminal truncation. This result implies that protein C-terminus may be more homogenous than protein N-terminus, which may have N-terminal truncation, signal peptide, propeptide, and subcellular localization sequence proceeding to the mature proteins[11]. We examined the tryptic peptides after the cleavage sites from protein sequences. Among them, 15 (about 48%) could not be identified if N-terminomics approaches were used. This is because the resulting peptides from N-terminal labeling of the peptides after the cleavage sites have lengths not suitable for MS identification. This suggests that C-terminomics is a complementary approach for N-terminomics in the identification of protein cleavages.
We further used the DAVID bioinformatics resources[27]to explore the cell compartments and biological processes for proteins listed in Table 1. These proteins are significantly enriched in the cytosolic and intracellular compartments (Fig. 4B). Biological process analysis found that a few ribosomal proteins, such as 30S ribosomal protein S6 and 50S ribosomal protein L6, are enriched in the translation. Several proteins, such as alcohol dehydrogenase yqhD, catalase-peroxidase, glycine dehydrogenase, and NADH-quinone oxidoreductase subunit G (Table 1), are enriched in oxidation and reduction. These results indicate that proteolytic cleavages might play important roles in regulating protein synthesis and redox-coupled reaction.

Fig. 4 Bioinformatic analyses of proteins with internal cleavages obtained from E. coli using ProC-TEL profiling. (A) Distribution of the identified C-terminal peptides from E. coli lysate. The numbers are the identified peptides from each category. (B) Distribution of cell compartments of cleaved proteins analyzed by DAVID bioinformatics resources. The data were plotted against the log(1/P value). (C) The number of amino acids at P1 and P1’ position of the internal cleavages from peptides listed in Table 1.
By examining the amino acids at the cleavage sites, we also found that the cleavages prefer to certain amino acids prior to (P1) or after (P1’) the cleaved peptide bonds (Fig. 4C). Almost all the amino acids at P1 position (30/31) have small (Gly and Ala), aliphatic (Leu and Ile), or acidic (Asp and Glu) side chains. The amino acids at the P1’ position also have preference on amino acids with small, aliphatic, acidic, or basic side chains. Surprisingly, a significant number of internal cleavages (12/31) occur right before Pro. This phenomenon may be closely associated with the special secondary or tertiary structures Pro may adopt at the cleavage sites.More internal cleaved peptides are required in order to obtain statistically significant result about the structural information at the cleavage sites.
Since trypsin digestion is performed in gel, it would be a significant improvement if the CPY labeling can be performed in gel prior to sample digestion. In our experimental procedure, proteins are required to be methyl esterified under the condition with minimal water (50 mmol/L HCl in methanol). We tried to conduct the methylation step in gel but the methylation solvent did not swell gel pieces well. Therefore, we did not perform the labeling experiment in gel. If the sample preparation prior to the labeling can be performed in SDS-PAGE, it would significantly extend the application of this method in solving more important biological relevant problems.
In our experiments, we used SDS-PAGE to separate proteins and remove the extra reagents (such as biocytinamide and SDS) and then we used in-gel trypsin digestion to obtain peptide mixture. In principle, this procedure can be performed in solution. In order to do so, the extra biocytinamide and SDS must be removed prior to trypsin digestion. Protein precipitation was a commonly used technique for this purpose. However, our trial with protein precipitation and resolubilization in a high concentration of urea found that protein recovery rate was very low because of the small amount of protein used in our experiments. Therefore, we did not use the in-solution digestion procedure. If protein recovery can be improved during protein precipitation, in-solution digestion with a low concentration of urea and subsequent purification of biotinylated peptides would be a better procedure for the isolation of C-terminal peptides.
In this work, we used ProC-TEL and mass spectrometer to identify C-terminal peptides from mature proteins and internal proteolytic cleavages. The identification of a protein or a cleavage is based on the detection of a single peptide. In addition, the identified peptides contain a biocytinamide modification at their C-termini. Therefore, the requirement for mass accuracy must be high enough for the identification of a signal peptide with modification for a protein. High resolution mass spectrometers, such as MALDI-TOF, Q-TOF, Orbitrap, or Q Exactive, are recommended for peptide analyses in this work.
Although ProC-TEL has been demonstrated to be an effective approach for the identification of protein C-terminal peptides and internal cleavages, it also has its disadvantages. Similar to N-terminomics, some tryptic peptides derived from the C-terminus of cleavage sites may not be suitable for MS/MS identification. This shortcoming could partially be overcome by the application of other proteases such as Glu-C and Lys-C during sample preparation. From our experiment, we cannot completely rule out the possibility that the CPY-catalyzed biotinylation may have bias towards specific amino acids at the protein C-termini[18]. In addition, protein solubility may be affected by methyl esterification, especially for membrane proteins. This problem may be alleviated by the addition of solubilization reagents compatible with CPY labeling after methyl esterification. The efficiency of the labeling may also depend on the nature of the amino acid sequence at protein C-terminus[18].
3 Conclusion
Although there are several approaches available for the identification of proteolytic cleavages in N-terminomics, C-terminal labeling is an alternative strategy. We have improved a protein C-terminal labeling procedure, ProC-TEL, for efficient labeling and positive isolation of protein C-terminal peptides from a complex protein mixture. Besides the C-terminal peptides of the mature proteins, many previously unknown internal cleavages were identified in cell lysate. These proteolytic cleavagesare most probably occurred in cells due to the harsh condition in the sample preparation, which deactivates proteases. A large fraction of the newly identified cleavages could not be detected through the N-terminomics due to the extreme length of the resulting peptides. Bioinformatic analyses showed that proteolytic cleavages may play important roles in protein translation and redox-coupled reaction. This work demonstrated that C-terminal labeling is a complementary approach to N-terminomics for the identification of proteolytic cleavages in complex biological systems.
Acknowledgements
We are grateful to Yarong Wang at the mass spectrometry core facility in the Medical School of Soochow University for the assistance during the mass spectrometry analysis. MALDI-TOF experiments were carried out at the core facility of the College of Chemistry at Soochow University.
REFERENCES
[1] Ghrayeb J, Inouye M. Nine amino acid residues at the NH2-terminal of lipoprotein are sufficient for its modification, processing, and localization in the outer membrane of Escherichia coli. J Biol Chem, 1984, 259(1): 463–467.
[2] Ehrmann M, Clausen T. Proteolysis as a regulatory mechanism. Annu Rev Genet, 2004, 38: 709–724.
[3] Kaufmann SH, Desnoyers S, Ottaviano Y, et al. Specific proteolytic cleavage of poly (ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res, 1993, 53(17): 3976–3985.
[4] Deraz EM, Kudo Y, Yoshida M, et al. MMP-10/stromelysin-2 promotes invasion of head and neck cancer. PLoS ONE, 2011, 7(10): e25438.
[5] Littlepage LE, Sternlicht MD, Rougier N, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res, 2010, 70(6): 2224–2234.
[6] Shao S, Li Z, Gao W, et al. ADAM-12 as a diagnostic marker for the proliferation, migration and invasion in patients with small cell lung cancer. PLoS ONE, 2014, 9(1): e85936.
[7] Li Y, Chen T, Kuklina AS, et al. Circulating proteolytic products of carboxypeptidase N for early detection of breast cancer. Clin Chem, 2014, 60(1): 233–242.
[8] Ye B, Cramer DW, Skates SJ, et al. Haptoglobin-α subunit as potential serum biomarker in ovarian cancer: identification and characterization using proteomic profiling and mass spectrometry. Clin Cancer Res, 2003, 9(8): 2904–2911.
[9] Hartmann EM, Armengaud J. N-terminomics and proteogenomics, getting off to a good start. Proteomics, 2014, 14(23/24): 2637–2646.
[10] Mahrus S, Trinidad JC, Barkan DT, et al. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell, 2008, 134(5): 866–876.
[11] Xu G, Shin SB, Jaffrey SR. Global profiling of protease cleavage sites by chemoselective labeling of protein N-termini. Proc Natl Acad Sci USA, 2009, 106(46): 19310–19315.
[12] Timmer JC, Enoksson M, Wildfang E, et al. Profiling constitutive proteolytic events in vivo. Biochem J, 2007, 407: 41–48.
[13] Kim JS, Dai Z, Aryal UK, et al. Resin-assisted enrichment of N-terminal peptides for characterizing proteolytic processing. Anal Chem, 2013, 85(14): 6826–6832.
[14] Prudova A, auf dem Keller U, Butler GS, et al. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics, 2010, 9(5): 894–911.
[15] auf dem Keller U, Prudova A, Gioia M, et al. A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol Cell Proteomics, 2010, 9(5): 912–927.
[16] Doucet A, Kleifeld O, Kizhakkedathu JN, et al.Identification of proteolytic products and natural protein N-termini by terminal amine isotopic labeling of substrates (TAILS). Methods Mol Biol, 2011, 753: 273–287.
[17] van Damme P, Staes A, Bronsoms S, et al. Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat Methods, 2010, 7(7): 512–515.
[18] Xu G, Shin SB, Jaffrey SR. Chemoenzymatic labeling of protein C-termini for positive selection of C-terminal peptides. ACS Chem Biol, 2011, 6(10): 1015–1020.
[19] Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell, 2008, 134(4): 679–691.
[20] Schilling O, Barre O, Huesgen PF, et al. Proteome-wide analysis of protein carboxy termini: C terminomics. Nat Methods, 2010, 7(7): 508–511.
[21] Tanco S, Gevaert K, van Damme P. C-terminomics: targeted analysis of natural and posttranslationally modified protein and peptide C-termini. Proteomics, 2015, 15(5/6): 903–914.
[22] Berne PF, Schmitter JM, Blanquet S. Peptide and protein carboxyl-terminal labeling through carboxypeptidase Y-catalyzed transpeptidation. J Biol Chem, 1990, 265(32): 19551–19559.
[23] Lin S, Lowe CR. C-Terminal labeling of immunoglobulin G with a cysteine derivative by carboxypeptidase Y catalyzed transpeptidation. Anal Biochem, 2000, 285(1): 127–134.
[24] Shevchenko A, Tomas H, Havlis J, et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc, 2006, 1(6): 2856–2860.
[25] Xu G, Deglincerti A, Paige JS, et al. Profiling lysine ubiquitination by selective enrichment of ubiquitin remnant-containing peptides. Methods Mol Biol, 2014, 1174: 57–71.
[26] The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res, 2015, 43: D204–D212.
[27] Huang D, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc, 2009, 4(1): 44–57.
[28] Winther JR, Sorensen P. Propeptide of carboxypeptidase Y provides a chaperone-like function as well as inhibition of the enzymatic activity. Proc Natl Acad Sci USA, 1991, 88(20): 9330–9334.
(本文责编 陈宏宇)
