红外加热辅助研磨法合成呋喃衍生物的研究
2016-06-29李景华张华贺
李景华,张华贺,张 纯
(浙江工业大学 药学院,浙江 杭州 310014)
红外加热辅助研磨法合成呋喃衍生物的研究
李景华,张华贺,张纯
(浙江工业大学 药学院,浙江 杭州 310014)
摘要:研究了一种红外加热辅助固相研磨合成呋喃衍生物的方法,此法绿色无溶剂,方便易操作.通过筛选不同的酸性催化体系及反应条件,发现H2SO4(30%)/SiO2作为催化剂,无需任何溶剂,由1,4-二羰基化合物在红外加热(控温80 ℃左右)辅助固相研磨条件下,发生Paal-Knorr缩合反应,高效地合成出了一系列呋喃衍生物.同时验证,该方法适用于不同取代基的1,4-二羰基化合物的Paal-Knorr 缩合反应.此方法操作简单方便,条件温和可控,反应时间短(6~30 min),分离收率高(83%~100%),产物纯度高.
关键词:红外加热;研磨;Paal-Knorr反应;呋喃衍生物
呋喃及其呋喃衍生物是一类重要的杂环化合物,在有机合成、材料化学以及医药等方面发挥着重要作用,而且许多天然产物中都含有它的结构单元,具有广泛的生物活性.关于呋喃及其衍生物的合成方法有很多[1],而利用1,4-二羰基化合物在酸催化下的Paal-Knorr反应[2]直接合成呋喃的方法简单方便,是目前应用较广泛的一种.近年来,有许多关于该反应改进方法的文献报道,其中最受关注的方法不外乎利用微波辅助合成方法[3],环境友好的离子液体合成方法[4]以及水溶剂法[5].这些合成条件的改进在一定程度上减少了环境污染,但是仍然存在一些不可避免的弊端,如使用毒性有机溶剂,高温,反应时间长[4],使用昂贵催化剂,分离提取困难等等.
将红外加热辅助固相研磨的方法引入到呋喃衍生物的合成中,代替传统的合成方法,大大缩短了反应时间,温度上也较微波反应更加温和可控,同时用硫酸/硅胶作为催化剂,以固相反应[6-7]代替常规的溶剂法反应,减少溶剂的消耗,降低对环境的污染.该方法操作简便,高效节能,值得我们进行研究.
1实验部分
1.1仪器与试剂
德国Büchi B-540熔点仪,Bruker Model Avance III 500 MHz核磁共振仪(溶剂CDCl3,内标TMS),红外灯250 W,薄层色谱板为G60 F254硅胶板,催化剂用硅胶为普通柱层析200~300目硅胶经120 ℃活化处理0.5 h.
1.2红外加热辅助Paal-Knorr反应固相法合成呋喃衍生物的一般方法
1,4-二羰基化合物在红外加热辅助固相研磨下经Paal-Knorr反应制备呋喃衍生物的反应式为

将普通柱层析硅胶(200~300目)0.2 g,浓硫酸(用量基于1,4-二羰基化合物质量的30%)一起置于研钵内,研磨均匀后,加入1 mmol 1,4-二羰基化合物,继续研磨1 min,将研钵放在红外灯下静置6~30 min(调节温度在80 ℃左右).TLC跟踪反应,反应完成后,用CH2Cl2浸提(8 mL×3),提取液先后经饱和NaHCO3溶液洗涤,水洗涤,无水硫酸镁干燥后,蒸去溶剂,将粗品置于真空干燥器里干燥即得到呋喃衍生物IIa~IIq.多数产物已足够纯(如IIc~IIg,可以直接送测NMR),部分产物可以用C2H5OH重结晶来进一步提纯(如IIa, IIk~IIq).谱图数据如下:
2,5-二甲基呋喃-3,4-二羧酸甲酯(IIa):淡黄色晶体;mp: 59~61 ℃(文献值[8]: 62~63 ℃);1H NMR (500 MHz, CDCl3)δ: 2.44 (s, 6H), 3.83 (s, 6H).
2,5-二甲基呋喃-3,4-二羧酸乙酯(IIb):淡黄色液体[4];1H NMR (500 MHz, CDCl3)δ: 1.29 (t,J=7 Hz, 6H), 2.39 (s, 6H), 4.25 (q,J=7 Hz, 4H).
2,5-二甲基-3,4-二乙酰基呋喃(IIc):白色晶体;mp: 56~58 ℃(文献值[9]: 63~64 ℃);1H NMR (500 MHz, CDCl3)δ: 2.41 (s, 6H), 2.43 (s, 6H).
2-甲基-5-苯基呋喃-3-羧酸甲酯(IId):淡黄色晶体;mp: 52~54 ℃(文献值[10]: 55~56 ℃);1H NMR (500 MHz, CDCl3)δ: 2.64 (s, 3H), 3.84 (s, 3H), 6.87 (s, 1H), 7.25~7.28 (m, 1H), 7.35~7.39 (m, 2H), 7.62~7.64 (m, 2H).
2-甲基-5-苯基呋喃-3-羧酸乙酯(IIe):淡黄色液体[11];1H NMR (500 MHz, CDCl3)δ: 1.35 (t,J=7 Hz, 3H), 2.63 (s, 3H), 4.30 (q,J=7 Hz, 2H), 6.87 (s, 1H), 7.22~7.26 (m, 1H), 7.34~7.37 (m, 2H), 7.61~7.63 (m, 2H).
5-(4-氟苯基)-2-甲基呋喃-3-羧酸甲酯(IIf):白色晶体[12];mp: 90~92 ℃;1H NMR (500 MHz, CDCl3)δ: 2.64 (s, 3H), 3.85 (s, 3H), 6.80 (s, 1H), 7.05~7.09 (m, 2H), 7.58~7.61 (m, 2H).
5-(4-氟苯基)-2-甲基呋喃-3-羧酸乙酯(IIg):白色晶体[11];mp: 58~60 ℃;1H NMR (500 MHz, CDCl3)δ: 1.37 (t,J=7 Hz, 3H), 2.64 (s, 3H), 4.31 (q,J=7 Hz, 2H), 6.81 (s, 1H), 7.06~7.09 (m, 2H), 7.59~7.62 (m, 2H).
5-(4-羟基苯基)-2-甲基呋喃-3-羧酸甲酯(IIh):淡黄色晶体[13];mp: 116~118 ℃;1H NMR (500 MHz, CDCl3)δ: 2.63 (s, 3H), 3.85 (s, 3H), 5.27 (s, 1H), 6.72 (s, 1H), 6.85~6.87 (m, 2H), 7.51~7.53 (m, 2H);13C NMR (500 MHz, CDCl3)δ: 13.83, 22.68, 29.69, 51.39, 103.73, 114.95, 115.69, 123.25, 125.38, 151.86, 155.40, 158.18, 164.76.
1-(2-甲基-5-苯基呋喃-3-)乙酮(IIi):淡黄色晶体;mp: 52~54 ℃(文献值[14]: 51~55 ℃);1H NMR (500 MHz, CDCl3)δ: 2.45 (s, 3H), 2.66 (s, 3H), 6.84 (s, 1H), 7.26~7.30 (m, 1H), 7.37~7.41 (m, 2H), 7.63~7.65 (m, 2H).
1-(5-(4-氟苯基)-2-甲基呋喃-3-)乙酮(IIj):淡黄色晶体[15];mp: 90~91 ℃;1H NMR (500 MHz, CDCl3)δ: 2.45 (s, 3H), 2.66 (s, 3H), 6.78 (s, 1H), 7.07~7.10 (m, 2H), 7.60~7.63 (m, 2H).
2-甲基-5-苯基呋喃(IIk):淡黄色晶体;mp: 38~39 ℃(文献值[15]: 39~40 ℃);1H NMR (500 MHz, CDCl3)δ: 2.42 (s, 3H), 6.09~6.10 (m, 1H), 6.58 (d,J=3 Hz, 1H), 7.24~7.28 (m, 1H), 7.38~7.41 (m, 2H), 7.67~7.69 (m, 2H).
2-甲基-3-苯甲酰基-5-苯基呋喃(IIm):淡黄色油状物[14];1H NMR (500 MHz, CDCl3)δ: 2.60 (s, 3H), 6.80 (s, 1H), 7.24~7.28 (m, 1H), 7.36~7.39 (m, 2H), 7.46~7.49 (m, 2H), 7.55~7.58 (m, 1H), 7.64~7.65 (m, 2H), 7.83~7.84 (m, 2H).
2,5-二苯基-3-苯甲酰基呋喃(IIp):淡黄色晶体;mp: 71~73 ℃(文献值[16]: 75~77 ℃);1H NMR (500 MHz, CDCl3)δ: 6.92 (s, 1H), 7.34~7.39 (m, 4H), 7.42~7.48 (m, 4H), 7.54~7.57 (m, 1H), 7.77~7.83 (m, 4H), 7.92~7.94 (m, 2H).
6,6-二甲基-2-苯基-6,7-二氢苯并呋喃-4(5H)-酮(IIq):白色针晶;mp: 89~90 ℃(文献值[17]: 100~101 ℃);1H NMR (500 MHz, CDCl3)δ: 1.18 (s, 6H), 2.41 (s, 2H), 2.83 (s, 2H), 6.89 (s, 1H), 7.29~7.32 (m, 1H), 7.39~7.42 (m, 2H), 7.65~7.66 (m, 2H).
2结果与讨论
2.1催化体系及反应条件的筛选
以Iq的Paal-Knorr反应作为模型反应,我们考察了不同酸在红外加热辅助固相条件下对反应的催化效果(表1).不使用酸作催化剂的条件下,红外灯照40 min,TLC跟踪反应显示,反应几乎没有进行(表1中序号为10),而使用了酸作催化剂后,反应效率大大提高.根据我们之前的研究结果,SiO2与其他酸配合体系在固相研磨合成反应中具有理想的催化活性[18],为此,我们也将其应用到呋喃环的合成中(表1中序号为 2~7,11~15).比较了几组不同催化体系的催化效果后,结果显示,与KHSO4/SiO2,CH3COOH/SiO2,p-TSA/SiO2,CF3SO3H/SiO2,Sm(OTf)3/SiO2相比,H2SO4/SiO2的催化体系效果最佳,反应30 min,收率就可以达到90%.尽管CH3SO3H/SiO2作为催化剂也能达到令人满意的效果,但浓H2SO4更廉价易得.另外,H2SO4负载在硅胶上,只需通过简单的过滤分离、加热活化等简单操作,就可以实现催化剂的再生重复利用.

表1 不同催化条件下1,4-二羰基化合物的反应情况1)
注:1) 总反应条件: 1,4-二羰基化合物 (1 mmol), SiO20.2 g 和30% 催化性酸(催化性酸用量为1,4-二羰基化合物质量的30%)室温下研磨1 min,红外灯下静置(控制温度80 ℃左右)至反应完全;2) 催化性酸用量为1,4-二羰基化合物的质量之比;3) 红外灯下放置的时间;4) 甲醇作溶剂,回流;5) 室温下研磨放置.
Iq的Paal-Knorr反应式为

催化剂的用量对目标产物的收率也有显著的影响(表1中序号为11~14).当浓硫酸的用量为底物质量的30%时,催化效果较好,而使用过多的浓硫酸时,可能由于部分原料分解,使目标产物的收率反而降低(表1中序号为14).作为对照试验,我们还考察了室温研磨(表1中序号为15)以及使用甲醇作溶剂(表1中序号为8)条件下的反应情况,效果均不理想.在此基础上,我们建立了较优的合成条件,即H2SO4(30%)/SiO2催化下红外加热辅助固相研磨.
2.2H2SO4/SiO2催化不同反应底物合成呋喃衍生物
为了进一步扩展该合成呋喃衍生物的方法对其他1,4-二羰基化合物的适用范围,我们又考察了带有不同取代基的1,4-二羰基化合物的Paal-Knorr 缩合反应,在红外加热辅助固相研磨条件下,H2SO4(30%)/SiO2催化均能高效地合成出相应的呋喃衍生物.从表2结果可以看出:所有反应在6~30 min内即可完成并获得较高收率.1,4-二羰基化合物的R1, R2, R3, R4位上无论是接供电子基团还是吸电子基团,反应均可以顺利进行.由于位阻效应的影响,底物Ip和Iq(表2中序号为13,14)所需反应时间略长.此外,底物Ia, Ib, Ik和Im由于反应过程中有副产物生成,以至于目标产物的收率稍低.
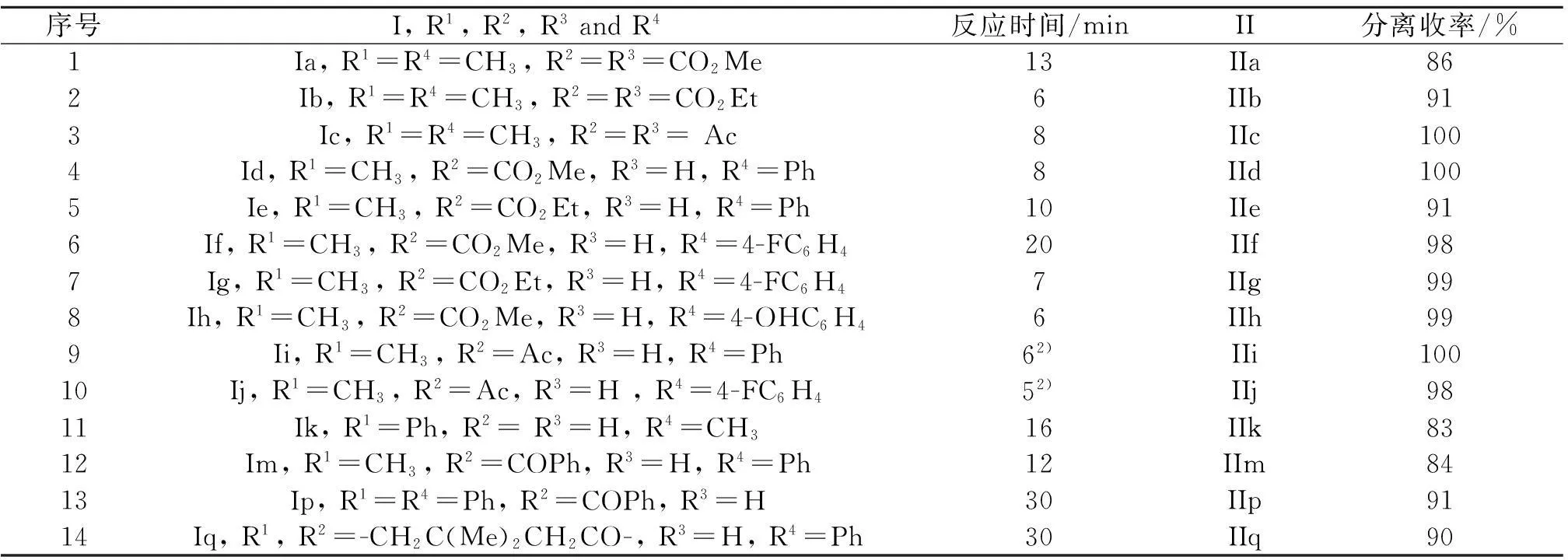
表2 红外加热辅助固相研磨法条件下的Paal-Knorr反应合成呋喃衍生物1)
注:1) 总反应条件: 1,4-二羰基化合物 (1 mmol), SiO20.2 g 和30% 浓硫酸(浓硫酸用量为1,4-二羰基化合物质量的30%)室温下研磨1 min,红外灯下静置(控制温度80℃左右)至反应完全;2) 室温下研磨.
3结论
在红外加热辅助固相研磨条件下,以H2SO4(30%)/SiO2为催化剂, 1,4-二羰基化合物发生Paal-Knorr反应合成呋喃衍生物.该方法与已报道的其他呋喃衍生物合成方法相比较,减少了有毒溶剂的使用,更加环保,产生的三废少;反应时间短(6~30 min),产品易分离,收率高,纯度高;H2SO4/SiO2体系作为催化剂,价格低廉,方便易得;红外加热固相研磨法与常规方法相比,干净、清洁.此红外加热固相法避免了已报道文献中的许多不足,为呋喃衍生物的合成提供了一种便捷高效的合成方法.
参考文献:
[1]XUA Haimei, ZHAO Huahua, SONG Huanling, et al. Functionalized ionic liquids supported on silica as mild and effectiveheterogeneous catalysts for dehydration of biomass to furanderivatives[J]. Journal of molecular catalysis a: chemical,2015,410:235-241.
[2]SOHEILA K, MAJID M H. Chapter three-Paal-Knorr reaction in the synthesis of heterocyclic compounds[J]. Advances in heterocyclic chemistry,2014,111:95-146.
[3]贾建洪,陈彬.微波在杂环类化合物合成中的研究进展[J].浙江工业大学学报,2009,37(1):36-43.
[4]WANG Gangqiang, GUAN Zhi, TANG Rongchang, et al. Ionic liquid as catalyst and reaction medium: a simple and efficient procedure for Paal-Knorr furan synthesis[J]. Synthetic communications,2010,40(3):370-377.
[5]WANG Gangqiang, GUAN Zhi, TANG Rongchang, et al. A simple preparation of ethyl 2,5-dimethylfuran-3-carboxylate and 2,5-dimethylfuran-3,4-dicarboxylic acid from diethyl 2,3-diacetylsuccinate[J]. Journal of heterocyclic chemistry,2009,46(3):540-543.
[6]张婷,徐时良,李景华.高苯丙氨酸的合成新工艺研究[J].浙江工业大学学报,2011,39(3):264-267.
[7]李郁锦,夏烈,高建荣.吩嗪类化合物的绿色合成研究[J].浙江工业大学学报,2013,41(3):286-288.
[8]CORT L A, RODRIGUEZ P A B. Some derivatives of naphthazarin and 2-methylnaphthazarin[J]. Journal of the chemical society[section] c: organic,1967(10):949-952.
[9]TREFIL'EV I A, LIFANOV E V. Oxonium compounds of the furan series[J]. Zhurnal obshchei khimii,1941,11:182-189.
[10]BRIONES J F, DAVIES H M L. Rh2(S-PTAD)4-catalyzed asymmetric cyclopropenation of aryl alkynes[J]. Tetrahedron,2011,67(24):4313-4317.
[11]DONG J J, ROGER J, POZGAN F, et. al. Low catalyst loading ligand-free palladium-catalyzed direct arylation of furans: an economically and environmentally attractive access to 5-arylfurans.[J]. Green chemistry,2009,11(11):1832-1846.
[12]HE Chuan, GUO Sheng, KE Jie, et al. Silver-mediated oxidative C-H/C-H functionalization: a strategy to construct polysubstituted furans[J]. Journal of the american chemical society,2012,134(13):5766-5769.
[13]PORRETTA G C, SCALZO M, CHIMENTI F, et al. Research on antibacterial and antifungal agents. III.Synthesis and antimicrobial activity of 2-methyl-5-aryl-3-furoic acids and 2-methyl-3-imidazolylmethyl-5-aryl furans[J]. Farmaco, edizione scientifica,1987,42(9):629-639.
[14]AOYAMA T, NAGAOKA T, TAKIDO T. One-pot synthesis of furans using base- and acid-supported reagents "Na2CO3/Al2O3-PPA/SiO2"[J]. Synthesis,2011(4):619-625.
[15]QIAN C Y. Synthesis of 2,3,5-trisubstituted furans by the acid-catalyzed decomposition of 1,2-dioxan-3-ols[J]. Journal of heterocyclic chemistry,1994,31(5):1219-1227.
[16]FRANCESCO B, STEFANIA R C, GIANLUCA M F, et al. A novel cyclization reaction between 2,3-bis(trimethylsilyl) buta-1,3-diene and acyl chlorides with straightforward formation of polysubstituted furans[J]. Chemical communications,2007,36:3756-3758.
[17]STAVROS K, SPYROS S. Photochemical reaction of phenyliodonium ylides of β-dicarbonyl compounds with terminal alkynes[J]. Journal of organic chemistry,1990,55(17):5041-5044.
[18]XU Shiliang, LI Chengping, LI Jinghua. Solid-state synthesis of β-enamino ketones from solid 1,3-dicarbonyl compounds and ammonium salts or amines[J]. Synlett,2009,5:818-822.
(责任编辑:刘岩)
Infrared heat aided grinding synthesis of furan derivatives
LI Jinghua, ZHANG Huahe, ZHANG Chun
(College of Pharmacuetical Sciences, Zhejiang University of Technology, Hangzhou 310014, China)
Abstract:A method for the synthesis of furan derivatives by infrared heat aided solid state grinding has been developed. The method can be operated conveniently and easily without solvent. Through screening of different acid catalysts and reaction conditions, a series of furan derivatives were synthesized from 1,4-diketones via solvent-free Paal-Knorr reaction catalyzed by H2SO4(30%)/SiO2 under infrared heat (the temperature about 80oC) aided solid state grinding conditions. This method is verified that it is suitable for the Paal-Knorr condensation reaction of 1,4-diketones with different substituents. The advantages of this method are simple procedure, mild conditions, rapidity(6~30 min), high isolated yields(83%~100%) and high purity.
Keywords:infrared heat; grinding; Paal-Knorr reaction; furan derivatives
收稿日期:2015-12-03
作者简介:李景华(1964—),男,浙江瑞安人,研究员,研究方向为绿色制药工艺技术,E-mail: lijh@zjut.edu.cn.
中图分类号:TQ251.1
文献标志码:A
文章编号:1006-4303(2016)03-0351-04
