功能化苯甲酰胺基-β-环糊精手性固定相的制备及其对黄烷酮对映体的拆分
2016-06-22林宇洲唐卫华
林宇洲, 周 杰, 唐 键, 唐卫华
(软化学与功能材料教育部重点实验室, 南京理工大学, 江苏 南京 210094)
功能化苯甲酰胺基-β-环糊精手性固定相的制备及其对黄烷酮对映体的拆分
林宇洲,周杰,唐键,唐卫华*
(软化学与功能材料教育部重点实验室, 南京理工大学, 江苏 南京 210094)
摘要:为了探讨功能基团对苯甲酰胺基-β-环糊精手性固定相手性拆分性能的影响,本文采用点击化学制备了两种手性固定相:全取代-4-氯-3-甲基苯甲酰氨基-6A-三唑基-β-环糊精键合硅胶手性固定相(CSP1)和全取代-5-氯-2-甲基苯甲酰氨基-6A-三唑基-β-环糊精键合硅胶手性固定相(CSP2)。利用核磁共振、红外光谱及元素分析等手段对固定相的结构进行了表征;在反相高效液相色谱条件下,通过对9种黄烷酮消旋体的拆分,对比研究了两种固定相的手性拆分性能。研究结果表明CSP1的拆分性能优于CSP2。这两种手性固定相仅在水与甲醇体系下即可实现对黄烷酮的手性拆分,展现了较好的应用前景。
关键词:高效液相色谱;环糊精;手性固定相;点击化学;黄烷酮;对映体拆分
采用纤维素或直链淀粉与苯异氰酸酯反应,进而制备的多糖类手性固定相是高效液相色谱(HPLC)应用中的一个研究热点[1],但此类手性固定相多为涂渍柱手性固定相,化学稳定性不高,对流动相溶剂的选择有严格的要求,因此限制了它们的应用。近年来,键合型手性固定相因其化学稳定性和溶剂耐受性而受到越来越多的研究者关注,比如功能纤维素通过Staudinger反应利用叠氮基还原键合硅胶[2],通过醚键键合功能化环糊精和硅胶[3,4]以及通过Gabriel反应氨基键合功能化环糊精和硅胶而制备的各类手性固定相[5]等。它们在HPLC应用中均展现了较好的分离效果,但是其制备过程的繁琐也是不争的事实。
环糊精(CD)因其独特的疏水空腔的包合作用、手性环境及结构可修饰性等优点,近年来在手性固定相开发上获得了很大的发展。点击反应因其反应选择性好、产率高及易操作性,在制备键合型环糊精固定相上展现了巨大潜力。其制备的环糊精固定相已成功应用于HPLC分析并且获得了优良的效果[6-11]。其中利用生成1,2,3-三唑结构的1,3-偶极环加成反应是应用最为广泛的键合方法,与前述反应相比,其反应步骤少,高效选择性极大地扩展了其应用,是现阶段发展迅速的实验方法之一。
近期,我们利用点击反应开发了环糊精和苯甲酰胺化环糊精手性固定相[7,8],在HPLC分析中取得了良好的手性分离效果。本文进一步报道了不同取代基修饰的苯甲酰胺化环糊精手性固定相的制备,并以黄酮类化合物对映体作为分析物,研究了两种手性固定相在RP-HPLC条件下的手性拆分性能;通过优化分离条件,探讨了此类色谱柱可能的拆分机理。
1实验部分
1.1仪器与试剂
安捷伦1260高效液相色谱分析仪,配G1315D二极管阵列检测仪(DAD)、G1329B四元泵、G1331C自动进样器、G1316A温度控制仪和安捷伦化学站数据处理软件(Version No. C.01. 04)(美国安捷伦公司); Nicolet iS-10傅里叶变换红外光谱(FT-IR)仪(美国赛默飞世尔科技公司); Vario EL-Ⅲ CHONS元素分析仪(德国Elementar Analysensysteme公司); Bruker AVANCE 500 MHz核磁共振(NMR)仪(美国布鲁克道尔顿公司)。
球形硅胶(粒径5 μm,平均孔径10 nm;瑞典Eka Chemicals公司); (3-氨基丙基)三乙氧基硅烷、丙炔酸、N,N-二环己基碳二亚胺、碘化亚铜、三苯基磷(分析纯,美国Sigma-Aldrich公司);甲醇、乙腈(色谱纯,美国Tedia公司);三氟乙酸、乙酸(色谱纯,上海百灵威科技公司);其余试剂及9种黄烷酮类消旋体(结构见图1)均购于上海安耐吉公司。实验所装色谱柱为定制不锈钢空柱(内径4.6 mm,长度250 mm(填料部分))。
1.2色谱固定相的制备
1.2.1炔基官能化硅胶的制备
炔基官能化硅胶的制备根据文献[12]进行。首先将球形硅胶(4 g)在120 ℃下真空干燥24 h使其活化,冷却至室温放置待用。在冰浴保护下依次向(3-氨基丙基)三乙氧基硅烷(5 mL)的无水二氯乙烷(30 mL)溶液中加入丙炔酸(1.5 mL)和N,N-二环己基碳二亚胺(4 mL),回流2 h;利用甲苯共沸、提纯,制备N-3-(三乙氧硅基)丙基-2-丙炔酰胺。在氮气保护下,将N-3-(三乙氧硅基)丙基-2-丙炔酰胺与活化硅胶(4 g)在甲苯(40 mL)中于120 ℃下搅拌回流24 h;经过滤、甲醇索氏提取24 h,即得目标硅胶。
1.2.2β-环糊精衍生物的合成
在氮气保护下,向双(三氯甲基)碳酸酯(三光气,纯度为99%,阿拉丁试剂)(10 g)的1,2-二氯乙烷(20 mL)溶液中滴入溶有4-氯-3-甲基苯胺(5 g)的1,2-二氯乙烷(20 mL)溶液;在80 ℃下回流24 h,经减压蒸馏获得4-氯-3-甲基苯基异氰酸酯。

图1 黄酮类对映体和环糊精点击手性固定相结构Fig. 1 Structures of 9 flavonoids and CD clicked chiral stationary phases (CSPs)
4-氯-3-甲基苯基异氰酸酯的表征:1H-NMR(500 MHz, CDCl3)δ值(ppm): 7.27~7.22 (5H-Ar, m, 1H), 6.95(2H-Ar, d, 1H), 6.85(6H-Ar, dd, 1H), 2.33(CH3, s, 3H)。
在氮气条件下,将所制备的4-氯-3-甲基苯基异氰酸酯或者5-氯-2-甲基苯基异氰酸酯(18.36 g)加入叠氮β-环糊精(3.8 g)[13]吡啶(20 mL)溶液中,在90 ℃下回流搅拌12 h。将减压蒸馏过的粗产品利用乙酸乙酯/石油醚(1∶10, v/v)混合溶剂重结晶提纯,即可获得全取代-4-氯-3-甲基苯甲酰氨基-6A-叠氮-β-环糊精或全取代-5-氯-2-甲基苯甲酰氨基-6A-叠氮-β-环糊精。
全取代-4-氯-3-甲基苯甲酰氨基-6A-叠氮-β-环糊精的表征:1H-NMR(500 MHz, d6-二甲基亚砜(DMSO-d6)δ值(ppm): 9.57(NH, d, 20H), 7.58~7.28(2H-Ar, m, 20H), 6.96(6H-Ar, s, 20H), 6.74(5H-Ar, d, 20H), 5.54~5.20 (m, 21H), 4.84(d, 7H), 4.34~3.93(m, 21H), 1.95(dd, 60H);13C-NMR(125 MHz, DMSO-d6)δ值(ppm): 153.5, 152.8, 138.2, 137.6, 136.9, 136.0, 135.3, 129.4, 128.7, 127.3, 121.5, 118.0, 71.9, 70.1, 63.6, 20.2, 19.9。
全取代-5-氯-2-甲基苯甲酰氨基-6A-叠氮-β-环糊精的表征:1H-NMR(500 MHz, DMSO-d6)δ值(ppm): 9.34~8.70(m, 20H), 7.91~7.71(m, 3H), 7.63~7.30(m, 25H), 7.11(dd, 22H), 6.81(dd, 30H), 5.43(d, 16H), 5.00(s, 4H), 4.36(d, 9H), 1.84(dd, 60H);13C-NMR(125 MHz, DMSO-d6)δ值(ppm): 153.8, 153.1, 144.2, 143.5, 137.5, 136.6, 131.6, 130.16, 129.8, 126.7, 124.2, 123.4, 71.2, 68.9, 63.2, 17.3, 16.3。
1.2.3衍生化环糊精键合硅胶手性固定相的制备
将炔基官能化硅胶(4 g)、衍生化β-环糊精(4 g)、三苯基磷(0.35 g)与无水二甲基甲酰胺(DMF)(40 mL)在80 ℃下搅拌48 h;经过滤、甲醇索氏提取纯化、干燥,即得全取代-4-氯-3-甲基苯甲酰氨基-β-环糊精键合硅胶手性固定相(CSP1)和全取代-5-氯-2-甲基苯甲酰氨基-β-环糊精键合硅胶手性固定相(CSP2)。
1.2.4色谱柱的装填
取衍生化环糊精键合硅胶(3 g),用甲醇作为匀浆液,使用压力泵在5.5 MPa压力下装入色谱柱管内,利用HPLC的流动相进行平衡,即得基于CSP1和CSP2的色谱柱。
1.3色谱条件和参数
配制黄烷酮消旋体样品的甲醇溶液(200 mg/L),储存于4 ℃冰箱中备用。HPLC分析中,进样量为10 μL,检测波长为220~280 nm,柱温为25 ℃,流速为1.0 mL/min。配制不同pH的三乙胺醋酸盐(TEAA, 1%(v/v))溶液,用醋酸调节所需要的pH。以甲醇为流动相,甲苯为标定物,25 ℃下流速为1 mL/min,计算CSP1和CSP2的柱效分别为10 794和10 130 塔板/m。手性拆分中,保留因子(k)、分离因子(α)和分离度(Rs)按照文献[14]方法进行计算。
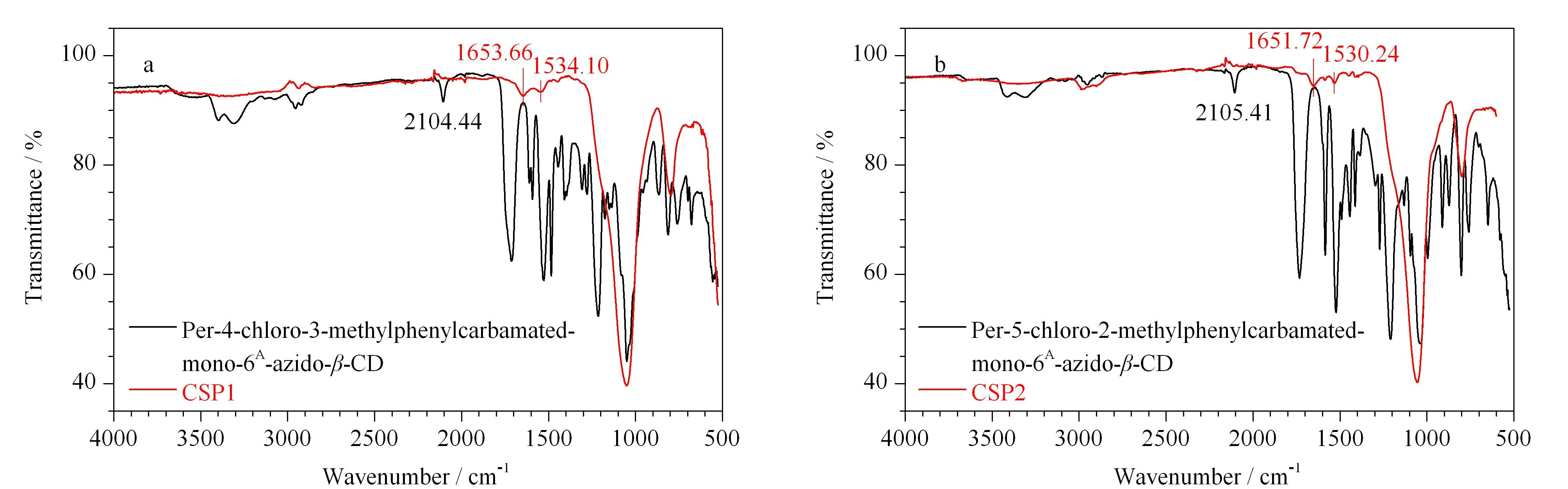
图2 (a)CSP1和(b)CSP2的红外光谱图Fig. 2 FT-IR spectra of (a) CSP1 and (b) CSP2
2结果与讨论
2.1色谱固定相的表征
两种衍生物的红外光谱图(图2红色)中3 317 和3 316 cm-1处可以看出存在分子内氢键作用,这对于增强改性环糊精的手性识别有特别的影响。在图2a中,1 653.66 cm-1对应于-C=O的特征峰,1 534.10 cm-1对应苯环的特征峰,且环糊精上叠氮基(-N3)的特征峰(2 104 cm-1)在CSP1的红外上消失,说明点击反应成功地将环糊精键合到硅胶上。图2b有类似的现象,无论是全取代-4-氯-3-甲基苯甲酰氨基-6A-叠氮-β-环糊精、全取代-5-氯-2-甲基苯甲酰胺基-6A-叠氮-β-环糊精,还是CSP1、CSP2,在谱图上有很大的相似性。

表 1 CSP1和CSP2对9种黄烷酮的手性拆分
Mobile phases: I, water-methanol (0∶100, v/v); II, water-methanol (10∶90, v/v); III, water-methanol (30∶70, v/v); IV, water-methanol (40∶60, v/v); V, methanol-water containing 1%TEAA (pH 4.0) (30∶70, v/v). TEAA: three ethylamine acetate.
元素分析测试表明,CSP1的C含量(质量分数,下同)为12.28%, N含量为0.822%, H含量为2.064%; CSP2的C含量为10.78%, N含量为0.35%, H含量为1.835%。两种CSP的C、N和H元素含量均比炔基化硅胶(C含量6.905%, N含量0.271%, H含量1.526%)有较大提高,说明环糊精衍生物成功点击键合在硅胶基质上。根据文献[14]中的计算方法,CSP1的负载率为0.21 μmol/m2, CSP2的负载率为0.18 μmol/m2。
固定相CSP1和CSP2虽然分子式相同,但苯甲酰胺基上的功能取代基的位置不一样。曾有课题组[15-17]探究过苯环上引入单一给电子基(-CH3)或吸电子基(-Cl),可有效改善未修饰苯环衍生化固定相的手性拆分性能。苯环的修饰可能提供额外的手性识别驱动力,进而提高固定相的手性分离能力。为此我们将吸电子基和供电子基选择性引入苯甲酰胺化手性固定相上,深入研究取代基的位置对手性拆分的影响,进而获得该类手性固定相的优化结构设计。
2.2色谱固定相的拆分性能
在RP-HPLC条件下,我们尝试了两种色谱柱对9种黄酮类化合物的手性分离。从表1的数据中能够初步判断出CSP1比CSP2对选取的黄酮类对映体有更强的拆分能力。在CSP1上,除橙皮素和柚皮素外,其余对映体在甲醇和水体系下便能得到基线分离,其中除7-羟基黄烷酮须在流动相含水的情况下才能基线分离外,剩下的6种对映体物质均能在纯甲醇作为流动相的条件下便能得到基线分离,其中3′-羟基黄烷酮获得了高达8.69的分离度。对CSP2而言,7-羟基黄烷酮、6-羟基黄烷酮以及橙皮素和黄皮素均不能得到拆分;对其他黄烷酮,可以通过调节水和甲醇流动相组成达到基线拆分。

图3 3种黄烷酮在CSP1和CSP2上的拆分效果比较Fig. 3 Enantioseparations of three racemates on CSP1 and CSP2 Mobile phase: water-methanol (20∶80, v/v).
为了对比两种固定相的效果差异,我们选取了在流动相为水/甲醇(20/80, v/v)条件下对黄烷酮、6-甲氧基黄烷酮、3′-羟基黄烷酮的拆分图进行对比。在图3中,我们能明显地看出CSP1的拆分效果优于CSP2,尽管CSP2的洗脱时间较短。选取的3种对映体在CSP1上均能达到基线分离,在CSP2上只有3′-羟基黄烷酮能达到基线分离。

图4 (a)CSP1和(b)CSP2在不同水含量的水/甲醇流动相下对黄烷酮的手性拆分Fig. 4 Enantioseparations of flavonoids on (a) CSP1 and (b) CSP2 with different contents of H2O in H2O/MeOH system as mobile phase
为了进一步探究CSP1和CSP2的拆分机理,我们研究了不同比例的水/甲醇流动相的拆分效果(见图4)。随着流动相中水含量的增加,两种固定相对黄烷酮的手性拆分性能有不一样的变化趋势。CSP2只可实现对5种样品的手性拆分,并且其分离度随着水含量的增加而呈现上升趋势。分离度提升的原因可由环糊精的拆分机理进行解释。在RP-HPLC条件下,环糊精手性拆分的驱动力主要有包合作用及其功能基团与对映体之间的相互作用力(如氢键作用、共轭作用、偶极-偶极作用)。包合作用同时受客体分子或单元的空间位阻影响。随着甲醇/水流动相中水含量的增加,一方面由于水的极性大于甲醇,可导致环糊精与对映体间作用力提高;另一方面由于甲醇可与对映体竞争进入环糊精空腔形成包合物,增加水含量可减少这种竞争,进而促进包合作用。这两方面的积极作用均可导致对映体与功能环糊精之间相互作用得以增强,手性识别能力提高,因此分离度提高。
但对于CSP1,黄烷酮的手性拆分与流动相的组成变化规律并不统一。不过,除橙皮素与柚皮素由于其芳香基团位阻较大,影响包合作用,其分离度未能达到基线分离(Rs=1.5)外,其他7种黄烷酮均获得了比CSP2上更为优异的手性拆分。除7-羟基黄烷酮外,6种黄烷酮的分离度在流动相水含量在0~40%范围内均大于3。在CSP1上,只有6-羟基黄烷酮和4′-羟基黄烷酮符合前述的解释,黄烷酮与7-羟基黄烷酮也能大致符合规律。但7-甲氧基黄烷酮、6-甲氧基黄烷酮和3′-羟基黄烷酮完全不符合前述规律,这意味着CSP1有着较为复杂的拆分机理。
对比9种黄烷酮的化学结构,能够发现3′-羟基黄烷酮在CSP1和CSP2上都有最高的分离度,β-环糊精的空腔适于包合萘类物质,因此认为黄酮类物质中的色酮环首先被包合于空腔中,黄酮上苯环的取代基与衍生化环糊精产生分子间作用力,产生识别机制,因此色酮环上含有取代基将不利于包合作用。在CSP2上,6-甲氧基黄烷酮和7-甲氧基黄烷酮的分离度较低,7-羟基黄烷酮和6-羟基黄烷酮不能拆分,可以解释为3′-羟基黄烷酮在两根色谱柱上有很高的分离度,符合假设,同时苯环上3位取代的空间位置有利于黄烷酮与环糊精产生更强的分子间作用力,相似的,色酮环6位的取代也利于产生分子间作用力。在CSP1和CSP2上,6-甲氧基黄烷酮的分离度高于7-甲氧基黄烷酮;在CSP1上,6-羟基黄烷酮的分离度优于7-羟基黄烷酮。甲氧基黄烷酮的甲基取代基为供电子基团,有利于黄酮与环糊精的共轭作用,因此甲氧基黄烷酮的分离相对优于羟基黄烷酮;在CSP1上,甲氧基黄烷酮的分离优于黄烷酮,说明空间位阻对环糊精与对映体的相互作用并不是决定性因素。

图5 CSP1在不同pH的甲醇-水流动相下对柚皮素的手性拆分Fig. 5 Enantioseparation of naringenin on CSP1 in MeOH/H2O (containing 1%(v/v) TEAA) (50∶50, v/v) with various pH values
硅胶因其表面的羟基而具有弱酸性,容易与碱性药物发生作用,使得以硅胶作为固定相的色谱柱在液相色谱分析时产生拖尾等多种不规则峰形,在流动相中添加三乙胺或其他物质往往能改善峰形,得到理想的结果。在对橙皮素和柚皮素的拆分过程中,发现单纯以水作为流动相的分离效果均不理想;但在流动相中添加三乙胺后,其分离因子和分离度在CSP1上均得到了提高;而CSP2仍然不能拆分它们。我们进一步考察了pH对柚皮素在CSP1上手性拆分的影响(见图5)。以甲醇/水(含1%(v/v)TEAA)(50/50, v/v)为流动相,结果表明,柚皮素在中性条件下几乎不能拆分;分离度在pH 5时达到最高,实现了完全基线分离,但保留时间较长;pH 4时,分析时间最短。橙皮素和柚皮素由于酚羟基的存在,具有弱酸性,pH可调节其离解性,在酸性条件下,其与环糊精间的相互作用力得到增强,因而获得了较好的拆分效果。同时,峰形在强酸性条件下得到较大改善。
3结论
本文通过点击反应成功制备了两种功能化苯甲酰胺基-β-环糊精手性固定相,利用高效液相色谱实现了对9种黄烷酮的手性拆分。全取代-4-氯-3-甲基苯甲酰胺基-β-环糊精固定相展现了较全取代-5-氯-2-甲基苯甲酰胺基-β-环糊精更好的手性拆分能力,对除橙皮素外的8种消旋体在较宽的流动相条件下全都实现基线分离。实验结果表明,在反相高效液相色谱中,环糊精固定相的手性拆分能力主要由包合作用、环糊精与对映体间相互作用力(如氢键作用、共轭作用、偶极作用等)决定。同时,流动相的极性调节可有效地改变两种环糊精固定相的手性拆分性能。更为重要的是,通过在苯环的间、对位上选择性引入供电子基团(如-CH3)和吸电子基团(如-Cl)修饰的苯甲酰胺基-β-环糊精手性固定相,可有效改善该类固定相的手性识别能力。本文的研究成果对于环糊精手性固定相的结构设计和应用具有很好的指导意义。
参考文献:
[1]Wang M. Chinese Journal of Chromatography, 2014, 32(2): 198
王敏. 色谱, 2014, 32(2): 198
[2]Tu H S, Fan J, Tan Y, et al. Chinese Journal of Chromatography, 2014, 32(5): 452
涂鸿盛, 范军, 谭艺, 等. 色谱, 2014, 32(5): 452
[3]Zhou Z, Li X, Chen X, et al. Anal Chim Acta, 2010, 678(2): 208
[4]Li X, Zhou Z M, Xu D, et al. Talanta, 2011, 84(4): 1080
[5]Lai X, Tang W H, Ng S C. J Chromatogr A, 2011, 1218(22): 3496
[6]Huang G, Ou J J, Zhang X D, et al. Electrophoresis, 2014, 35(19): 2752
[7]Pang L M, Zhou J, Tang J, et al. J Chromatogr A, 2014, 1363(10): 119
[8]Zhang S P, Wang H H, Tang J, et al. Anal Methods, 2014, 6(7): 2034
[9]Xu X F, Shen A J, Guo Z M, et al. Chinese Journal of Chromatography, 2013, 31(3): 185
徐雪峰, 沈爱金, 郭志谋, 等. 色谱, 2013, 31(3): 185
[10]Zhang Y P, Guo Z, Ye J X, et al. J Chromatogr A, 2008, 1191(1/2): 188
[11]Fan Q, Zhang K, Tian L W, et al. J Chromatogr Sci, 2014, 52(5): 453
[12]Wang Y, Xiao Y, Yang T, et al. Tetrahedron Lett, 2008, 49(35): 5190
[13]Tang W H, Ng S C. Nat Protoc, 2008, 3(4): 691
[14]Siles B A, Brian H H, Dorsey J G. J Chromatogr A, 1995, 704(2): 289
[15]Kahina S A, Fairouz T, Badjah-Hadj-Ahmed A Y. Anal Bioanal Chem, 2009, 395(2): 507
[16]Fan Q, Zhang K, Tian L W, et al. J Chromatogr Sci, 2014, 52(5): 453
[17]Lin C, Fan J, Liu W N, et al. J Pharmaceut Biomed, 2014, 98(9): 221
Preparation of phenylcarbamoylated β-cyclodextrin chiral stationary phases and the enantioseparation of flavonoids
LIN Yuzhou, ZHOU Jie, TANG Jian, TANG Weihua*
(Key Laboratory of Soft Chemistry and Functional Materials, Ministry of Education,Nanjing University of Science and Technology, Nanjing 210094, China)
Abstract:Cyclodextrin (CD) based chiral stationary phases (CSPs) have simulated great attention due to their unique chiral recognition ability. Functionalized cyclodextrins bonded silica gel as chiral stationary phases have been dramatically developed due to their chemical stability and solvent tolerability. To explore the functionalization of phenylcarbamoylated β-cyclodextrin CSPs on their enantioselectivities, 4-chloro-3-methylaniline and 5-chloro-2-methyl phenyl isocyanate were employed. Two CSPs have been thus developed by clicking the CD derivatives onto alkynylated silica support. They include per-4-chloro-3-methylphenylcarbamoylated β-cyclodextrin clicked CSP (CSP1) and per-5-chloro-2-methylphenylcarbamoylated β-cyclodextrin clicked CSP (CSP2), which have both electron-donating (methyl) and withdrawing (chlorine) groups in the phenylcarbamate moieties, but different locations. The CSPs were successfully characterized in terms of structure using nuclear magnetic resonance (NMR), Fourier transform infrared spectroscopy (FT-IR) and elementary analysis. Their enantioselectivities wereevaluated in reversed-phase high performance liquid chromatography (HPLC). The comparison study on the enantioseparation of nine flavonoids with the two CSPs demonstrates the higher enantioselectivities of CSP1 over CSP2, because of the different locations of electron-donating (methyl) and withdrawing (chlorine) groups in the phenylcarbamate moieties of CD derivatives. The baseline enantioseparations achieved in water/methanol as mobile phase offered great potential for the CSPs to be used in practical application.
Key words:high performance liquid chromatography (HPLC); cyclodextrin (CD); chiral stationary phase (CSP); click chemistry; flavonoids; enantioseparation
DOI:10.3724/SP.J.1123.2015.06038
*收稿日期:2015-06-23
基金项目:国家自然科学基金项目(21305066).
中图分类号:O658
文献标识码:A
文章编号:1000-8713(2016)01-0096-07
色谱手性分离专刊·研究论文
*通讯联系人.E-mail:whtang@mail.njust.edu.cn.
Foundation item: National Natural Science Foundation of China (Grant No. 21305066).
