团簇分子[Zn-(CH3CH2OH)n]2+的荧光光谱研究及理论光谱验证
2016-06-15吴晓静蒋卫国于亚鹏余学会程龙玖
吴晓静, 蒋卫国, 于亚鹏, 余学会, 程龙玖
1. 合肥工业大学化学与化工学院, 安徽 合肥 230009
2. 安徽大学化学化工学院, 安徽 合肥 230601
团簇分子[Zn-(CH3CH2OH)n]2+的荧光光谱研究及理论光谱验证
吴晓静1, 蒋卫国1, 于亚鹏1, 余学会1, 程龙玖2
1. 合肥工业大学化学与化工学院, 安徽 合肥 230009
2. 安徽大学化学化工学院, 安徽 合肥 230601
通过荧光光谱实验和理论计算对金属离子在乙醇溶液中的微团簇结构进行了研究。 荧光光谱实验发现在275~330 nm范围存在较强的乙醇分子荧光峰, 当加入盐离子(Zn2+)后该处荧光强度逐渐变弱, 说明盐的加入对乙醇体系的荧光效率产生了影响, 破坏了原有乙醇分子之间的作用, 使得其刚性结构发生改变, 荧光效率降低, 同时Zn2+与乙醇分子通过溶剂化作用形成了新的微团簇, 在350~380 nm之间出现了新峰。 通过对团簇结构[Zn-(H2O)n]2+(n=1~3)进行优化比较, 得到了相对精确及运算成本较低的密度泛函理论B3LYP方法, 并应用于Zn2+在乙醇溶液中团簇结构计算。 结果表明体系的第一溶剂化层存在着n=1~6的微团簇分子, 且最多为6。 比较理论计算荧光光谱与实验荧光光谱, 进一步说明了Zn2+与乙醇形成了新的微团簇及计算方法的可行性。
溶剂化; 密度泛函; 团簇分子
引 言
金属离子在溶液中溶剂化现象非常普遍, 其在生物化学等方面起着重要的作用。 关于金属离子在溶液中的团簇结构一直吸引着研究者的关注, 如近年来科研工作者对水溶液中的离子微观结构的研究[1-2], Science[3]杂志更是对不同浓度溶液的微观团簇作了特别的报道。 随着科技的发展许多关于金属离子溶剂化的理论计算得以实现[4-5], 尽管光谱学被应用于探究盐溶液的微观结构如红外, 拉曼等[6-7], 直接的实验证明仍然有待进一步的研究。 前期工作主要涉及了轻元素(如Li+, Na+, Ca2+等), 重元素(如La3+等)在溶液中的微观结构[8-9]。 过渡金属在现代生物学如酶的活性[10], 工业[11], 科技应用[12]等领域都有着不可替代的地位。 其中Zn2+是人体必需营养元素之一, 对人的成长发育, 内分泌, 免疫等许多生理及病理过程起着十分重要的作用[13]。
乙醇作为常见的有机溶剂, 表面活性剂[14], 猝灭剂[15], 有着广泛的应用, 但是关于液态乙醇团簇结构的研究进展仍然非常缓慢。 Sarkar等[16]曾经利用X射线衍射对常温下的乙醇进行了研究, 发现由于分子间氢键的连接, 液态乙醇中存在四聚物、 五聚物、 六聚物的分子团簇。 本研究通过量子化学计算方法和荧光实验来研究金属离子与溶剂之间的相互作用。 通过实验荧光光谱与理论计算荧光光谱, 研究了Zn2+在乙醇溶液中的微观结构, 发现Zn2+与CH3CH2OH形成了新的团簇分子, 且比单纯的乙醇团簇分子更稳定, 验证了理论计算的可行性。
1 荧光光谱
1.1 仪器及药品
乙醇(>99.9%,色谱纯)、 六水合硝酸锌,试剂没有进一步纯化。 分别配置成浓度为0.510, 0.357, 0.255, 0.153 mol·L-1的溶液。
实验选用日本日立公司F-2700型分子荧光光谱仪测定, 光源为150 W氙灯, 测量环境温度为(24±1)℃, 石英比色皿, 测量范围为220~600 nm, 扫描精度为0.5 nm, 扫速为60 nm·min-1。 实验中先固定一个荧光发射波长, 测得激发光谱曲线, 从中得出不同激发波长, 固定最佳激发光波长值, 测得对应的荧光发射光谱曲线。
1.2 结果及分析
实验测得不同浓度梯度的荧光光谱, 如图1所示。 金属离子浓度梯度从a—e依次增加, 其中a为色谱纯乙醇。 实验中固定的激发波长为253 nm, 由于电子从激发态回到基态的过程中往往伴随着电子的辐射, 振动弛豫, 内转换, 即会产生比发射波长大的荧光, 图中在275~330 nm波段间出现的较强的波峰为乙醇分子的荧光峰。
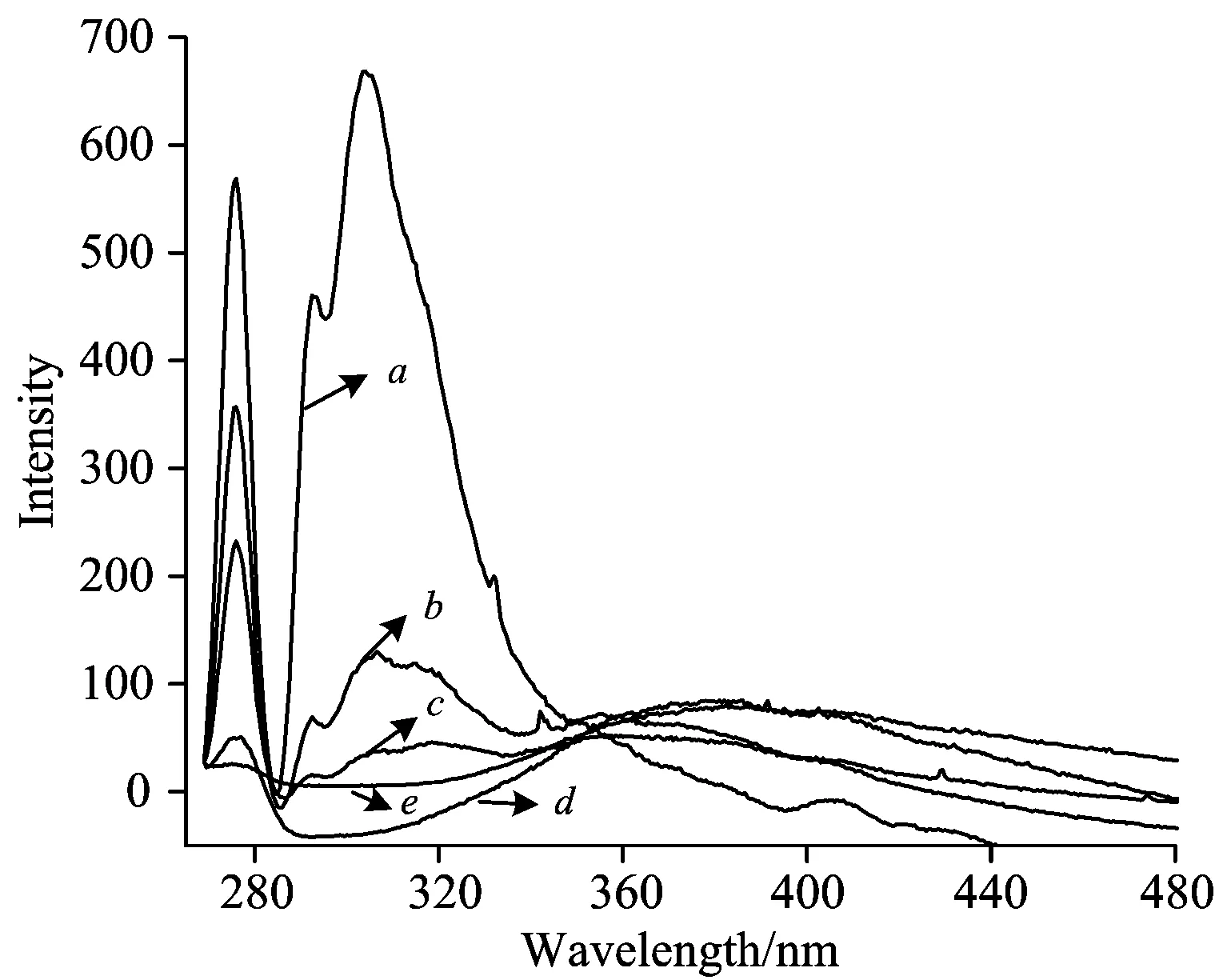
图1 不同浓度的Zn2+/乙醇在253 nm的激励光下的荧光光谱图
Fig.1 Fluorescent spectra of Zn2+/ethanol solution with different concentrations induced by 253 nm
由乙醇的分子式可知, 乙醇分子中含有杂原子基团C—O—H, 该基团除了成键电子外, 氧元素上还有两对孤对电子, 这种电子在紫外光下容易被激发而形成n→σ*跃迁, 故乙醇在近紫外区有吸收 , 激发态电子不稳定, 跃迁回到基态, 产生荧光。 另外, 在色谱纯乙醇分子体系中乙醇分子之间通过氢键缔合成链状结构, 形成大的刚性团簇, 增加了荧光效率。
从图1可以看出随着盐浓度的增加在275~330 nm处的荧光强度变弱, 说明了Zn(NO3)2的加入对乙醇体系的荧光效率产生了影响, 破坏了原有乙醇分子之间的缔合作用, 刚性结构发生改变, 盐离子的加入同时增加了乙醇分子之间的碰撞效率, 进而增加了乙醇分子之间的无辐射跃迁几率, 导致荧光效率降低。 同时在350~380 nm之间出现了新峰, 这可能是由于加入的Zn2+与乙醇分子通过溶剂化作用形成了新的微团簇, 且随着盐离子浓度的进一步增加在275~330 nm处的荧光峰强度逐渐减小趋于零, 比350~380 nm出现的荧光峰要弱, 这是由于Zn2+与乙醇分子形成的新团簇分子比单纯的乙醇团簇更加稳定。
2 理论计算
2.1 [Zn -(H2O)n]2+团簇构型的理论计算及结果分析
为了得到快速准确的计算方法, 首先对结构较简单的H2O作为溶剂进行了计算。 分别采用ab initio(HF, MP2, CCSD)以及DFT(B3LYP, B3PW91, LSDA)对团簇分子[Zn-(H2O)n]2+(n=1~3)进行结构优化计算。 计算过程中采用“6-31G(d,p)+LanL2DZ”混合基组, 用关键词GEN对基组进行设定, 其中对H和O元素选取6-31G(d,p)基组, 对Zn2+采用Lanl2dz赝势基组以减少对金属优化的计算量并且赝势基组包含了对金属相对论效应的修正[17], 所有计算除非特殊说明, 均采用该混合基组。
CCSD方法被普遍认为是在计算电子结构中最为精确的方法[18], 此外CCSD方法在预测键长以及团簇分子之间的相互作用能等方面有很高的准确性[19], 虽然CCSD方法有着精确的计算, 但是其计算成本较高、 耗时较长(如用CCSD, HF, B3LYP, MP2优化[Zn-(H2O)3]2+的CPU TIME分别为17 h 21′48″, 40″, 20′42″, 30′14″), 而且几乎不适合用来计算n>3时的团簇分子[20]。 计算中选用不同的泛函方法对团簇[Zn-(H2O)n]2+(n=1~3)的结构优化并同时计算其频率, 将生成各个团簇的能量变化ΔE(m)(m=B3LYP, B3WP91, LSDA, MP2, HF), 与ΔE(CCSD)相比较, 得到能量的绝对误差, 即|ΔE|=|ΔE(m)-ΔE(CCSD)|, 如图2所示。 从图2中可以看出B3LYP, MP2, HF在计算能量变化方面与CCSD相比绝对误差相对较小。
图2 不同的方法得到的能量变化与CCSD方法比较的绝对差值; |ΔE|
Fig.2 The absolute energy error values of different methods compare to CCSD method; |ΔE|
另外所得Zn—O之间的键长如表1所示, 当n=1~2时, 相比于HF方法MP2与B3LYP方法所得Zn—O之间的键长更为准确, 其中MP2更接近CCSD方法, 当n=3的时候也是MP2与B3LYP较为准确, B3LYP方法得到的Zn—O更接近CCSD方法, 所以可认为MP2与B3LYP较为准确, 同时考虑到计算成本, 在计算n=4~6更复杂的体系时B3LYP的计算时间相比于MP2大大缩短, 故采用B3LYP来对团簇构型进行优化。
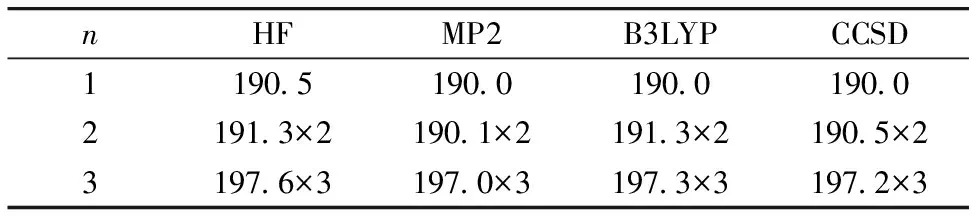
表1 不同方法优化团簇分子[Zn-(H2O)n]2+(n=1~3)
2.2 [Zn-(CH3CH2OH)n]2+结构的理论计算结果及分析
通过上述比较, 选择计算成本较低的B3LYP泛函方法, 以及“6-31G(d,p)+LanL2DZ”混合基组, 应用于Zn2+与结构较为复杂的溶剂(CH3CH2OH)的团簇构型。 优化结果[Zn-(CH3CH2OH)n]2+(n=1~7)如图3所示。
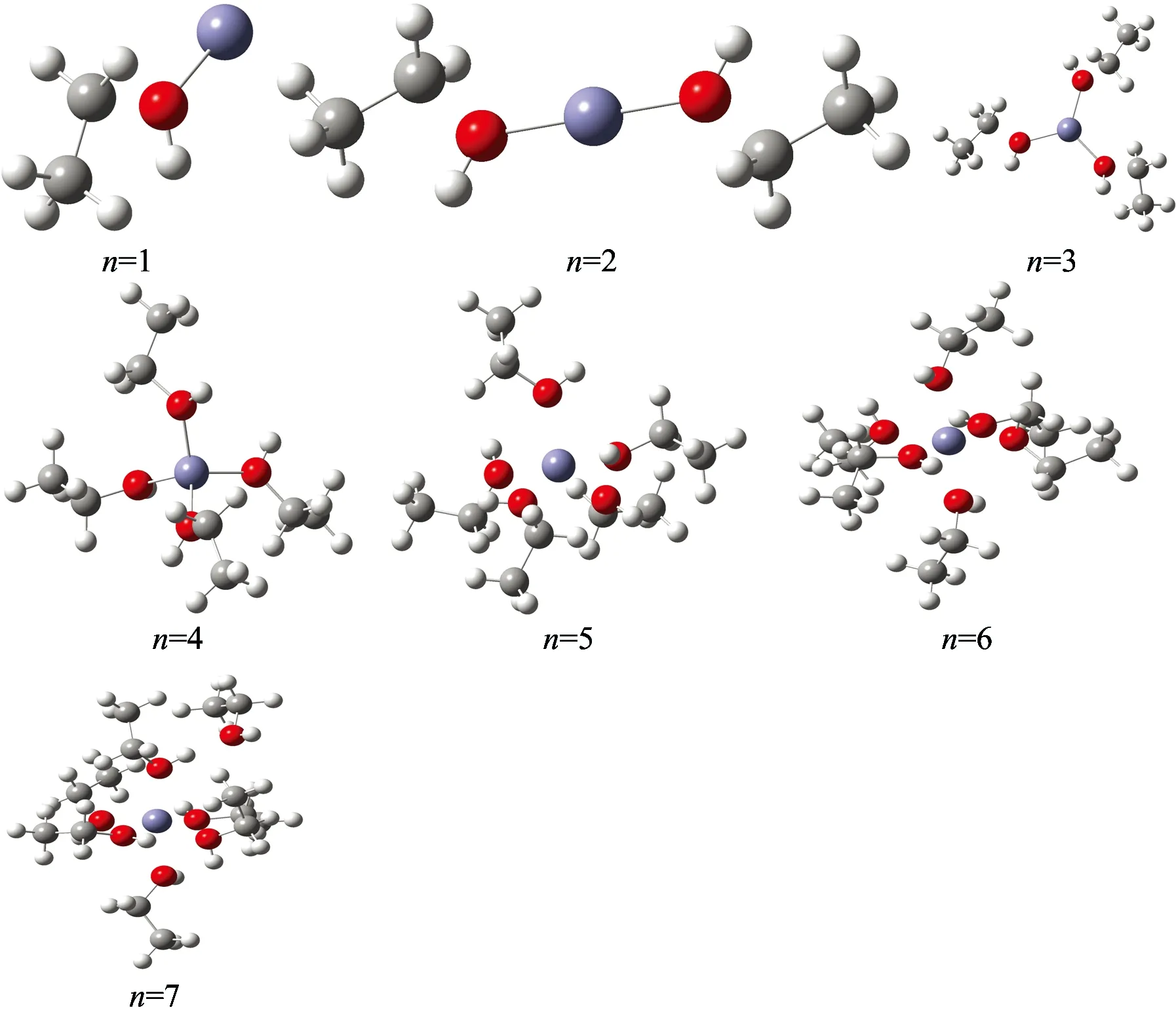
图3 B3LYP优化得到盐溶液中的可能团簇结构
Fig.3 B3LYP optimized the possible structures of [Zn-(H2O)n]2+clusters(n=1~7)
由图3可知当n>6时[Zn-(CH3CH2OH)n]2+的团簇会出现第二极化层, 这是由于空间位阻导致第一溶剂化层容纳不下更多的乙醇分子。 由于第二溶剂化层乙醇分子与阳离子之间的距离较远, 阳离子对乙醇分子的吸引力较弱, 所以实验主要研究第一溶剂化层的团簇分子的性质。
Zn2+与CH3CH2OH形成微团簇结构的过程如下
[Zn-(CH3CH2OH)n-1]2++
CH3CH2OH=[Zn-(CH3CH2OH)n]2+n=1~6
在298.15 K, 101.325 KPa条件下微团簇[Zn-(CH3CH2OH)n]2+的热力学参数见表2。 随着n的增加体系的能量逐渐降低, 当n=6时, 团簇体系的能量最低, 当n>6时, 团簇分子形成次外层。
图4为反应的吉布斯自由能的变化(ΔG), 当n=1~6的时候ΔG<0, 说明每个团簇分子都可以通过自发反应生成, 并且随着乙醇分子数目的增加最终可能生成[Zn-(CH3CH2OH)6+m]2+(m是次外层的乙醇分子数目)的团簇分子。
表2 团簇[Zn-(CH3CH2OH)n]2+(n=1~6)的热力学参数
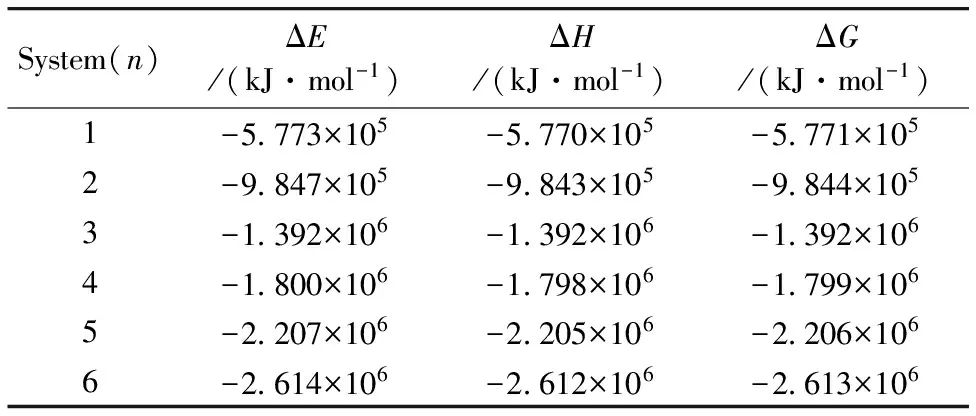
Table 2 Thermodynamic parameters of [Zn-(CH3CH2OH)n]2+
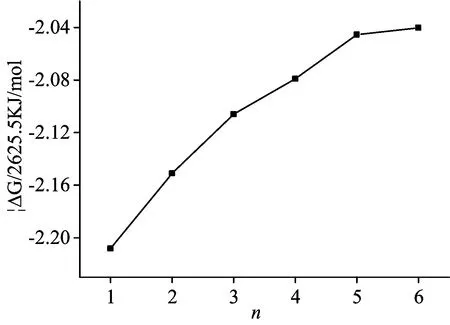
图4 [Zn-(CH3CH2OH)n]2+(n=1~6)团簇的吉布斯自由能(ΔG)变化
Fig.4 The changes of Gibbs(ΔG)of [Zn-(CH3CH2OH)n]2+(n=1~6)clusters
2.3 理论荧光光谱计算
为了验证光谱实验和理论计算的可行性, 对[Zn-(CH3CH2OH)n]2+(n=1~6)微团簇分子进行了发射光谱计算, 在已优化好的团簇构型基础进行如下计算; 首先通过Time-dependent density functional method(TD-DFT)对优化的结构进行垂直激发态计算得到吸收光谱, 其次确定需要计算的激发态以及激发态的个数, 实验中计算的是第一激发态(Root=1), 激发态数目是15(nstates=15), 最后对上述激发态进行结构优化和频率计算得到了不同团簇对应的理论中心发射波长(λ), 能量(E)以及振荡强度(f), 见表3。 可以看出理论微团簇峰主要集中在339.02~414.63 nm之间, 与350~380 nm之间Zn2+/乙醇溶液出现新峰的实验荧光光谱较吻合, 如图5所示, 其中图5(a)为实验光谱, 图5(b)为计算光谱。
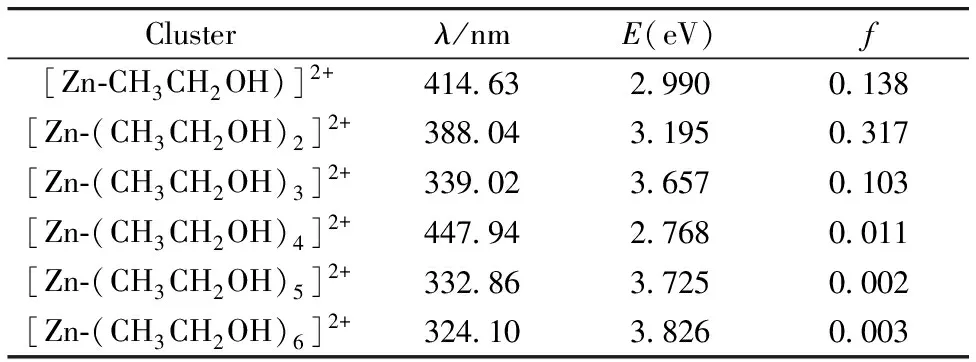
表3 不同团簇的理论发射光谱(λ)及震荡强度(f)
图5中1~6条曲线分别对应的是n=1~6时的荧光光谱曲线, 7号曲线是这六个光谱的累计峰曲线,e是盐溶液浓度最高的荧光光谱实验谱线。 比较图5中理论计算光谱与实验光谱可以看出, 加入Zn2+之后理论计算得到的荧光谱峰与实验的荧光谱峰基本相同, 且理论与实验得到的光谱在波长380 nm左右处荧光强度最大, 这进一步表明了Zn2+的加入对分析纯无水乙醇的荧光产生了影响, 使得原有乙醇体系的刚性结构被破坏, 乙醇分子与金属离子(Zn2+)产生了六种新的微团簇分子。
此外, 在稀溶液中, 物质的荧光强度与物质的浓度成正比例关系, 结合表3与图5可以发现, 当n=1~3的时候理论的荧光强度较大,n=4, 5, 6时强度很弱, 说明溶液中的

图5 实验荧光光谱及理论计算可能团簇的荧光光谱
微团簇结构主要以n=1~3的形式存在。
3 结 论
通过采用不同的计算方法对Zn2+在水溶液中的可能团簇结构进行优化, 比较得出B3LYP是相对精确及运算成本较低的方法, 应用于Zn2+-乙醇中微团簇结构的优化计算, 结果表明乙醇的第一溶剂化层最多只能容纳6个乙醇分子。 通过理论计算得到的热力学参数, 可知n=1~6的团簇分子在溶液中都是可能存在的。
荧光光谱实验也较好地验证了模拟计算, 溶剂化的作用产生了新的微团簇分子, 导致其荧光光谱出现了新峰。 与实验光谱比较, 理论计算荧光光谱确定了各种微团簇峰的位置, 其荧光发射强度表明微团簇分子[Zn-(CH3CH2OH)n]2+主要以n=1~3的形式存在。
致谢: 作者非常感谢日本福冈大学理学院Toshio Yamaguchi教授在实验和计算过程中给予的建议和帮助。
[1] Spezia R, Beuchat C, Vuilleumier R, et al. The Journal of Physical Chemistry B, 2012, 116(22): 6465.
[2] Amaro-Estrada J I, Maron L, Ramírez-Solís A. Physical Chemistry Chemical Physics, 2014, 16(18): 8455.
[3] K Mueller, J Yeston. Science, 2011, 331: 1491.
[4] Matsumiya M, Kamo Y, Hata K, et al. Journal of Molecular Structure, 2013, 1048: 59.
[5] Monti D, Jónsson E, Palacín M R, et al. Journal of Power Sources, 2014, 245: 630.
[6] Karabacak M, Bilgili S, Mavis T, et al. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2013, 115: 709.
[7] Terashima Y, Takeda K, Honda M. Chemical Physics, 2014, 430: 23.
[8] WU Xiao-jing, DAI Yun(吴晓静, 代 云). Science China: Chemistry(中国科学: 化学), 2011, 41(10): 1597.
[9] WU Xiao-jing, QUAN Jun-jie, YU Ya-peng, et al(吴晓静, 权俊杰, 于亚鹏, 等). Chemistry(化学通报), 2013, 76(10): 935.
[10] Wilson M, Hogstrand C, Maret W. Journal of Biological Chemistry, 2012, 287(12): 9322.
[11] Shi Z, Zhang C, Tang C, et al. Chemical Society Reviews, 2012, 41(8): 3381.
[12] Finn C, Schnittger S, Yellowlees L. J. Chem. Commun., 2012, 48: 1392?1399.
[13] Lin S, Lin X, Yang Y, et al. Aquaculture, 2013, 406: 79.
[14] Zdziennicka A, Jańczuk B. Journal of Colloid and Interface Science, 2011, 354(1): 396.
[15] Rastogi A, Al-Abed S R, Dionysiou D D. Applied Catalysis B: Environmental, 2009, 85(3): 171.
[16] Sarkar S, Joarder R N. J. Chem. Phys., 1994, 100: 5118.
[17] K Laasonen, Pasquarello A, Car R, et al. Phys. Rev. B, 1993, 47(16): 10142.
[18] Bukowski R, Szalewicz K, Groenenboom G C A. Science, 2007, 315: 1249.
[19] Feller D, Peterson K A. J. Chem. Phys., 2007, 126: 114105.
[20] Ayala R, Martinez J M, Pappalardo R R. Journal of Physical Chemistry B, 2008, 112(17): 5416.
[Zn-(CH3CH2OH)n]2+Clusters Probed with Fluorescence Spectroscopy and Computation
WU Xiao-jing1, JIANG Wei-guo1, YU Ya-peng1, YU Xue-hui1, CHENG Long-jiu2
1. School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei 230009, China
2. College of Chemistry & Chemical Engineening, Anhui University, Hefei 230601, China
In this paper the cluster structures of [Zn-(CH3CH2OH)n]2+have been investigated with spectroscopic experiment and theoretical calculation. According to the fluorescence spectroscopy experiments, the fluorescence peak of ethanol molecules was found between 275~330 nm. A new peak appeared between 350~380 nm after the metal ions (Zn2+) was added into ethanol solution due to the generation of new clusters of molecules, and the original fluorescence peak of ethanol molecules became weak owing to the destroyed structure of ethanol molecules induced by Zn2+. The cluster structures of Zn2+in water solution were investigated by using different methods. By comparing the results, a more accurate and fast B3LYP method of DFT was found and applied to optimize the possible structures of [Zn-(CH3CH2OH)n]2+. The results suggested that the first solvation shell of the system is up to six ethanol molecules, and thermodynamic parameters also shows the six kinds of molecular clusters which are likely in the solution. Moreover compared the theoretical fluorescence spectroscopy with experimental fluorescence spectroscopy, new clusters [Zn-(CH3CH2OH)n]2+have been generated, with [Zn-(CH3CH2OH)n]2+(n=1~3) as main constructions.
Solvation; DFT; Clusters of molecular
Feb. 12, 2015; accepted Jun. 20, 2015)
2015-02-12,
2015-06-20
国家自然科学基金面上项目(21273008)资助
吴晓静, 1963年生, 合肥工业大学化学与化工学院副教授 e-mail: wuxiaojing@ustc.edu
O645; O641
A
10.3964/j.issn.1000-0593(2016)08-2527-05
