芳乙酸类化合物的合成研究进展II
2016-06-06陈叶童毛春飞陈继漆郑土才
陈叶童,徐 欢,杨 杰,毛春飞,陈继漆,李 见,魏 斌,郑土才,*
(1.衢州学院化学与材料工程学院,浙江 衢州 324000;2.江苏迪安化工有限公司,江苏 灌南 223500)
笔者在第1部分叙述了零碳取代芳烃与2个及以上碳原子单元连接法合成芳乙酸类化合物的研究进展[1]。本文接着介绍单碳基团取代芳烃与1个及以上碳原子单元的直接或间接连接法合成芳乙酸类化合物的研究进展。
2 单碳基团取代芳体烃与1个及以上碳原子单元连接法
2.1 甲基芳烃与1个及以上碳原子单元反应
2.1.1 直接羧化反应和直接酯化、水解反应
芳烃环上甲基受吸电子基团活化,被强碱夺去质子形成碳负离子,直接与二氧化碳发生亲核加成、酸化得到芳乙酸。形成的碳负离子也可以与氯甲酸酯或碳酸酐等发生缩合得到芳乙酸酯,水解得到芳乙酸:

Shabanov等报道了1种由钠、氯苯、甲苯和催化剂直接反应制备苯乙酸的方法,钠与氯苯先反应生成苯基钠,再与甲苯反应产生苄基碳负离子,与干冰直接羧化得到苯乙酸,收率92%[2]。甲苯的芳环缺少强吸电子基团,需要使用苯基钠那样的强碱,反应条件较为苛刻,不适用于有敏感基团存在的甲基芳烃。
Salman等报道,4-甲基水杨酸以溴乙烷(EtBr)、碳酸钾、二甲基亚砜(DMSO)进行乙基化和酯化,再以正丁基锂(n-BuLi)、二异丙胺(DIPA)、四氢呋喃(THF)、1,3-二甲基四氢嘧啶-2-酮(DMPU)处理,通入二氧化碳羧化得到糖尿病治疗药瑞格列奈中间体3-乙氧基-4-乙氧羰基苯乙酸,总收率67%[3]:

仇章明等报道4-甲基水杨酸以溴乙烷、碳酸钾、N,N-二甲基甲酰胺(DMF)进行乙基化和酯化,再以n-BuLi、DIPA、2-甲基四氢呋喃处理,通入二氧化碳羧化得到3-乙氧基-4-乙氧羰基苯乙酸,总收率61.5%[4]。钱珊等也以4-甲基水杨酸为原料,经与溴乙烷/碳酸钾反应得2-乙氧基-4-甲基苯甲酸乙酯,再在二异丙胺基锂(LDA)、六甲基磷酰胺(HMPA)作用下生成相应的苄基碳负离子,与二氧化碳直接羧化得到3-乙氧基-4-乙氧羰基苯乙酸,产率73%[5]。
类似地,Aswathanarayanappa等报道,4-甲基-2-乙氧基苯甲酸乙酯以n-BuLi、DIPA、THF处理,再与叔丁氧碳酸酐反应得到3-乙氧基-4-乙氧羰基苯乙酸叔丁酯,收率达81%[6]。王迷娟等以4-甲基水杨酸为原料,经硫酸二乙酯(Et2SO4)同时酯化和醚化得到2-乙氧基-4-甲基苯甲酸乙酯,再经LDA作用生成碳负离子,与氯甲酸乙酯羧化(ClCOOEt)、NaOH水解得到3-乙氧基-4-乙氧羰基苯乙酸,总收率58%[7]:

Zheng等报道了1种1-氯-6-甲氧基异喹啉-3-醇的合成新法,2-甲基-4-甲氧基苯腈在LDA、THF、DMPU的作用下,低温通入二氧化碳羧化得到2-氰基-5-甲氧基苯乙酸,收率82%,再经氯化亚砜氯化、盐酸催化环合得到目标产物[8]。
2.1.2 芳烃活泼甲基与草酸酯缩合、断裂氧化反应
与2.1.1节类似,芳烃环上活泼甲基脱去质子形成的碳负离子,也可以与草酸酯发生缩合得到芳基丙酮酸酯,再经水解、断裂氧化生成芳乙酸:

蔡可迎等以邻硝基甲苯为原料,在乙醇钠作用下与草酸二乙酯缩合,然后经水解、双氧水断裂氧化得到邻硝基苯乙酸,收率62%;进而以FeO(OH)或氢氧化钠快速沉淀CuCl2和AlCl3的混合液制得的催化剂CuO-Al2O3催化水合肼还原得到邻氨基苯乙酸,最后经重氮化卤置换反应得到邻卤苯乙酸[9-10]。
郭希等报道,2-硝基-4-氯甲苯先与钠/乙醇反应,再与草酸二乙酯缩合得到2-硝基-4-氯苯丙酮酸乙酯,水解、双氧水断裂氧化得到2-硝基-4-氯苯乙酸,以2-硝基-4-氯甲苯计总收率40%,再经硝基还原、环合、傅克酰化制得新型抗精神病药齐拉西酮中间体5-(2-氯乙酰基)-6-氯吲哚酮[11]。
孙平华等以2-甲基-3-硝基苯乙酸为原料,经酰氯化、胺化、还原得N,N-二丙基-2-甲基-3-硝基苯乙胺。该中间体再与乙醇钠、草酸二乙酯缩合,收率73%;水解、双氧水断裂氧化得到2-硝基-6-[2-(二丙胺基)乙基]苯乙酸,收率81%;最后经还原、缩合等反应合成抗帕金森氏症药盐酸罗匹尼罗[12]。
2.2 卤化苄与1个碳原子单元反应法
2.2.1 卤化苄的直接羰基化和羧基化反应
卤化苄在钴、钯等配合物催化下与一氧化碳、水进行羰基化合成芳乙酸是研究得较多的一种芳乙酸合成方法。卤化苄还可以与二氧化碳通过电解还原羧化,合成芳乙酸,避免了价格昂贵的羰基化催化剂及剧毒的一氧化碳:

Qiu等详细研究了氯化苄经羰基化合成苯乙酸的多种影响因素,包括催化剂的种类和含量、溶剂、加料方式、反应温度、氢氧化钠和三苯基膦含量、表面活性剂、有机相与水相的相比等,得到最优催化剂为Pd(PPh3)2Cl2,优化条件下苯乙酸收率高达97.6%[13]。李剑利等报道了同一反应,以Co(PPh3)2Cl2为主催化剂,短链2-烷基-1-二(2-羟乙基)-2-咪唑啉氯化物为相转移催化剂,但苯乙酸收率仅为30%左右[14]。然而,金洗郎等报道了同一反应,也是以Co(PPh3)2Cl2为主催化剂,短链2-烷基-1-二(2-羟乙基)-2-咪唑啉氯化物为相转移催化剂,苯乙酸收率达70%左右[15]。
王立中等以邻氯氯苄为原料,八羰基二钴为催化剂,氢氧化钠和一氧化碳共同作用下直接羰基化合成邻氯苯乙酸。考察了不同反应条件对收率的影响,优化反应条件为:常压下,醇水体积比为2:1,氢氧化钠的质量分数30%,催化剂与邻氯氯苄的量比1:40,反应温度50℃,产率达74.5%[16]。
钟建新等、何人宝等报道2,4,5-三氟氯苄与一氧化碳在催化剂四羰基钴盐存在下进行羰基化反应,得到2,4,5-三氟苯乙酸,收率89%[17-18]。
Jones等报道了氯苄类化合物经钯催化的羰基化合成芳乙酸及其酯类化合物的研究,催化剂为2-溴甲基苯甲醇、2-溴和2-碘苯甲醇、2-碘苯乙醇衍生的有机钯配合物[19]。
王萍等报道了对二甲苯经氯甲基化和催化羰基化合成2,5-二甲基苯乙酸的工艺,其中羰基化以Pd(PPh3)2Cl2、Pd(PPh3)4或 Pd(PPh3)2(OAc)2为催化剂,三乙胺或吡啶、叔戊醇或异丙醇存在下进行,收率87%[20]。
Isse等报道氯化苄类化合物在二氧化碳饱和的乙腈中在银阳极发生电催化还原性羧化,得到苯乙酸类化合物,氯苄、对三氟甲基氯苄和对甲氧基氯苄均得到收率很高的对应的苯乙酸[21]。
陆嘉星等以对氯甲基苯乙烯为原料,在四丁基氟硼酸铵和DMF中,在常压以二氧化碳饱和后恒电流电解,后处理得到对乙烯基苯乙酸[22]。
2.2.2 卤化苄的格氏试剂羧化反应
卤化苄与镁在THF等非质子性溶剂中反应生成格氏试剂,再与干冰或二氧化碳反应、水解得到芳乙酸:

这种方法是经典的芳乙酸合成方法之一,但反应条件要求较高,芳环上不能带有能与格氏试剂反应的官能团如羟基、羰基等。
吴永兰等先以氯苄与镁、THF反应得到格氏试剂,与二氧化碳加成得到苯乙酸,进一步与对苯二酚在BF3-Et2O、DMF、PCl5中反应合成6-羟基异黄酮[23]。
祝宏等以联苯为原料,经氯甲基化得氯甲基联苯,再经格氏试剂与二氧化碳加成、酸化生成联苯乙酸,以联苯计总收率53.2%[24]。
张群辉等报道了一种2,4,5-三氟苯乙酸的合成,2,4,5-三氟氯苄在引发剂存在和有机溶剂中与镁反应得到格氏试剂,通入二氧化碳反应,水解得到2,4,5-三氟苯乙酸,收率70%以上[25]。冯秀娟等报道了类似的工艺,2,4,5-三氟氯苄或溴苄在引发剂1,2-二溴乙烷存在和有机溶剂如THF或甲基叔丁基醚中与镁反应得到格氏试剂,加入固体干冰反应,水解得到2,4,5-三氟苯乙酸,收率接近定量[26]。
2.2 .3 卤化苄的氰化、水解反应法
卤化苄与金属氰化物(MCN)经亲核取代反应生成芳乙腈,水解得到芳乙酸:

该法是芳乙酸的经典合成方法之一,氰化反应一般需要使用相转移催化剂(PTC)。缺点是氰化物剧毒,水解要控制条件使腈完全转化为酸。
为了开发苯乙酸的绿色制备工艺,石超君和吕秀阳系统的研究了近临界水中苯乙腈的无催化水解反应动力学。结果表明,苯乙腈水解反应为典型的连串反应,苯乙酰胺为中间产物,且能高选择性地得到最终产物苯乙酸。并以一级连串反应动力学模型对实验数据进行了拟合,得到了苯乙腈、苯乙酰胺水解反应活化能分别为64.4、82.4 kJ/mol[27-28]。该工艺反应过程无需加任何催化剂,解决了酸、碱催化水解的污染,过程简单、绿色,产品纯度和收率高。
傅杰等考察了不同二氧化碳添加量对高温液态水中苯乙腈水解的影响,计算了不同二氧化碳添加量和不同温度对高温液态水反应体系pH的影响。结果表明,在473.2 K,0、0.2、0.4 MPa二氧化碳压力下的水解反应速率常数分别为0.62×10-3、0.41×10-3、0.30×10-3min-3,而相对应的pH分别为5.6、4.1、3.9[29]。对于碱催化机理为主导的苯乙腈水解反应,二氧化碳的加入不但不能促进苯乙腈在高温液态水中的水解,反而会降低苯乙腈的水解速率,加入量越大,影响越大。
针对高温液态水中苯乙腈无催化水解制苯乙酸反应速度慢的缺点,任浩明和吕秀阳又提出了含氨高温液态水中苯乙腈水解制备苯乙酰胺和苯乙酸的方法,并研究了该条件下苯乙腈水解反应的动力学。动力学拟合结果表明,氨的加入主要提高了苯乙腈水解产生苯乙酰胺的速度,而对苯乙酰胺进一步水解产生苯乙酸的影响相对较小。当氨加入量为229.8 mg/L时苯乙腈和苯乙酰胺水解反应活化能分别降低至57.2、67.7 kJ/mol-,因而氨的加入有可能改变了苯乙腈水解反应的途径。所得苯乙酰胺和苯乙酸可以通过调节pH加以分离[30-31]。由于氨易于回收,因此该方法具有简单、环境友好的特点。
吴毅等以对氯苯乙腈为原料,经四丁基溴化铵(TBAB)催化的质量分数30%氢氧化钠水解得到对氯苯乙酸,收率78.0%;再经N-溴代丁二酰亚胺(NBS)溴代和微波辅助碳酸钠水解制得农业杀菌剂双炔酰菌胺中间体4-氯-α-羟基苯乙酸[32]。
Baelen等研究了对氯苯乙腈和3,4-二甲氧基苯乙腈在微波作用下的氢氧化钾/水/甲苯两相水解反应。对氯苯乙腈在微波功率50 W、160℃反应30 min,相应酸的收率为92%。3,4-二甲氧基苯乙腈在微波功率50 W、160℃反应8 h,相应酸的收率为80%,还有8%的酰胺及2%未反应的腈[33]。
郭新艳等以3,4-二甲氧基苯乙腈为原料,经氢氧化钠、水、甲醇水解得到3,4-二甲氧基苯乙酸,收率83%,再经酰化、环化等得到天然产物番荔枝宁[34]。
朱玉松等以3,5-二甲氧基苄溴为原料,在无水乙醇中与NaCN反应生成3,5-二甲氧基苄腈,收率96%。再在氢氧化钠、水、甲醇作用下,水解得到3,5-二甲氧基苯乙酸,收率82%。再经Knoevenagel、Perkin等反应合成白藜芦醇类似物[35]。
陈勇等将苯乙腈以浓硝酸、浓盐酸硝化制得4-硝基苯乙腈,Fe粉还原得到4-氨基苯乙腈,再在TBAB催化下,在C5H11ONO/Cu作用下,加噻吩偶联得到4-(2-噻吩基)苯乙腈,最后在氢氧化钠、水、乙二醇中水解得到4-(2-噻吩基)苯乙酸,总收率82%[36]。
Vincze等报道了5-羟基-4-甲氧基-2-溴苯乙腈经氯苄、碳酸钾的酚羟基苄基化和氢氧化钠、水、乙醇水解、酸化得到5-苄氧基-4-甲氧基-2-溴苯乙酸,水解收率71%[37]。
陈康等以2,4-二氯苄氯为原料,在TBAB催化下进行氰化,再以氢氧化钠水解得到杀虫杀螨剂螺螨酯中间体2,4-二氯苯乙酸,总收率84.8%[38]。
邹建平等报道,对二甲苯经甲醛、浓盐酸的氯甲基化得到2,5-二甲基氯苄,收率83.6%;以TBAB催化氰化钠水溶液的氰化得到2,5-二甲基苯乙腈,收率97.0%;最后以硫酸水溶液水解得到杀虫杀螨剂螺虫乙酯中间体2,5-二甲基苯乙酸,收率96.1%[39]。
李惠等以均三甲苯为原料,在甲醛、浓盐酸作用下氯甲基化制得2,4,6-三甲基苄氯,再与氰化钠在十六烷基三正丁基溴化铵(CTBAB)相转移催化下氰化制得2,4,6-三甲基苯乙腈,2步收率83%。氢氧化钠水解制得杀虫杀螨剂螺螨甲酯中间体2,4,6-三甲基苯乙酸,收率94.3%[40]。刘宇等以均三甲苯经甲醛、浓盐酸的氯甲基化得到2,4,6-三甲基氯苄,不经任何处理,直接以CTBAB催化氰化钠水溶液的氰化得到2,4,6-三甲基苯乙腈,两步收率88.7%,最后以硫酸水溶液水解得到2,4,6-三甲基苯乙酸,收率97.6%[41]。宋尚海等以2,4,6-三甲基苯乙腈为原料,在TBAB催化下经碱性水解得到2,4,6-三甲基苯乙酸,收率约80%[42]。
蔡正艳等以1,3,5-三异丙苯为原料,在多聚甲醛、盐酸作用下经氯甲基化得到2-氯甲基-1,3,5-三异丙基苯,再在TBAB的催化下,与氰化钾经氰化反应得到2,4,6-三异丙基苯乙腈,最后氢氧化钠水解得到降血脂药阿伐麦布中间体2,4,6-三异丙基苯乙酸,3步总收率95%[43]。
但飞君等以胡椒醛为原料,与甲醛、氢氧化钠经Cannizzaro反应得到3,4-亚甲基二氧苄醇,收率99.2%。浓盐酸氯化得到3,4-亚甲基二氧苄氯,收率95%。在十六烷基三甲基溴化铵(CTMAB)的催化下,氰化钠氰化得到腈,收率85.9%。以氢氧化钠在1-乙氧基-2-甲氧基乙烷、水中水解得到3,4-亚甲二氧基苯乙酸,收率85.6%。五氯化磷去保护得到3,4-二羟基苯乙酸[44]。
贺新等以3,4,5-三甲氧基苯甲醛为起始原料,经TBAB、硼氢化钾、水还原,氯化亚砜氯化,氰化钠、TEBAC、水、甲苯氰化,氢氧化钠水解“一锅”反应得到3,4,5-三甲氧基苯乙酸,总收率64.2%[45]。
孙晋瑞等以4-苯基苄基氯为原料,经亚铁氰化钾、醋酸铜、DMF氰化及酸性水解制得联苯乙酸,总收率82%。其中氰化剂亚铁氰化钾价廉、无毒,条件温和,避免了剧毒的氰化物,具有较好的前景[46]。
Salem等在新颖5α-还原酶抑制剂合成中,报道了4-甲基-4’-苯氧基二苯酮以NBS、过氧化苯甲酰在四氯化碳中溴化,氰化钠、二氧六环氰化和质量分数40%氢氧化钠水解,酸化合成4-(对苯氧基苯甲酰)苯乙酸,4步收率分别为44%、47%和63%[47]。
Lee等报道2-、3-或4-苄氧基氯苄经氰化钠、DMF氰化,稀硫酸、甲醇水解得到相应的苄氧基苯乙酸,氰化和水解收率均在96%以上[48]。
Nakhostin等报道,2-邻氟(或氯)苯基-4-氯苯甲酸经氯化亚砜、乙醇酯化,氢化铝锂、THF还原,氯化亚砜、吡啶、苯氯化,氰化钠、DMSO氰化生成相应的2-邻氟(或氯)苯基-4-氯苯乙腈,再经氢氧化钾、乙醇或丁醇的水解,酸化生成2-邻氟(或氯)苯基-4-氯苯乙酸(或2-邻氯苯基-4-氯苯乙酸)[49]。
李燕等、钟建新等、刘一超等报道了1,2,4-三氟苯和多聚甲醛及氯化剂反应得到2,4,5-三氟苄氯,再经氰化、水解得到抗糖尿病药西他列汀中间体2,4,5-三氟苯乙酸,其中氯化剂包括氯磺酸、氯化氢/硫酸、氯化钠/硫酸、盐酸/氯化锌和三氯化磷/醋酸,氰化一般采用氰化钠/PTC/乙醇/水、氰化亚铜/N-甲基-2-吡咯烷酮(NMP)、氰化钠或氰化钾/离子液体如1-丁基-3-甲基咪唑四氟硼酸盐等,水解可使用盐酸/醋酸、硫酸/醋酸、或氢氧化钠等,收率可达99%[50-52]。该工艺产品含量高,合成路线短,条件温和。
每日优鲜的前置仓基本在100平米左右,在SKU的选择上采取精选模式,每日优鲜只精选不到1000个商品,大型超市的SKU数量在2万左右,天猫、京东的生鲜数量也能在4000个左右,所以每日优鲜SKU的数量还不足超市的1/20。因为分散的小面积前置仓容纳不下太多的商品数量,超过数量的SKU在小仓中是分拣不出来的,这样一来顾客可供选择的余地太小了。缩减SKU,在短期内可以提高流通效率,但从长期来看,其实是将用户推向拥有更丰富选择的竞争对手。
卤化苄与金属氰化物经亲核取代生成芳乙腈,进而水解为芳乙酸的方法报道较多,其中卤化苄可以通过芳烃的氯甲基化、甲基芳烃的自由基卤化、芳醛、芳酸及其衍生物的还原、卤化,或者转化为磺酸酯代替卤化苄。氰化反应一般使用氰化钠、氰化钾或氰化亚铜,前2种氰化物的氰化一般在醇、水的均相,或甲苯、水的两相体系中进行,添加PTC可以加快反应,提高收率。氰化亚铜氰化一般在强极性非质子性溶剂如DMF、DMSO、NMP等中进行。氰化物剧毒,影响了这些反应的工业化应用。水解反应可以采用碱性水解或酸性水解,重点是如何控制反应完全转化为酸。
2.3 芳醛与1个及以上碳原子单元反应法
芳醛可以通过多种方法转化为芳乙酸,其中经还原、卤化或磺酸酯化、氰化、水解的4步反应法,是最常用的实验室方法之一,已在2.2.3节中讨论。
2.3.1 与硝基甲烷的缩合、氧化反应
Matt等发现,伯烷基硝基化合物、伯溴代烃以及伯醇甲磺酸酯在亚硝酸钠、醋酸和DMSO的作用下,可以转化为相应的羧酸,其中2-苯基-1-硝基乙烷、2-苯基-1-溴乙烷和2-苯基-1-乙醇甲磺酸酯分别得到收率为95%、85%和45%的苯乙酸,反应条件十分温和。溴代烃和甲磺酸酯被认为是在反应条件下先转化为硝基烷烃的。2-芳基-1-硝基乙烷很容易由芳醛与硝基甲烷经Knoevenagel缩合、选择性还原烯键得到,也可以由硝基甲烷与氯苄类化合物经硝基甲烷碳的烷基化获得[53]。
许慧等以3,4-二甲氧基苯甲醛为原料,在醋酸铵、醋酸作用下与硝基甲烷经Knoevenagel缩合得到3,4-二甲氧基-β-硝基苯乙烯,收率88.6%,硼氢化钾选择性还原烯键得到3,4-二甲氧基-(β-硝基乙基)苯,收率85.7%,再经亚硝酸钠、醋酸和DMSO作用得到罂粟碱、维拉帕米、贝凡洛尔等药物中间体3,4-二甲氧基苯乙酸,收率82.5%[54]:


Nikalje等报道,芳醛与硝基甲烷经类似羟醛缩合反应即Henry反应生成1-芳基-2-硝基乙醇,然后在Cu(II)盐催化和30%的醋酸、甲醇水溶液中反应转化为α-氧代芳乙酸:

醋酸铜的催化效果优于硫酸铜或氯化铜。其中对甲基、对甲氧基、对氯取代苯的衍生物,3,4,5-三甲氧基、3-硝基-4-甲基取代苯的衍生物,2-萘基和2-呋喃基的衍生物均能获得较好的效果,收率59%~93%[55]。α-氧代芳乙酸可经已知方法还原为芳乙酸,见1.1.2节[1]。硝基甲烷与芳醛的Henry反应易于进行、条件温和、收率良好,该方法具有一定的实用价值。
2.3.2 经转化为扁桃酸、还原脱羟基反应

邓旭忠等以3,4,5-三甲氧基苯甲醛为原料,在氯仿、氢氧化钠和PTC作用下制得3,4,5-三甲氧基扁桃酸,收率71.7%,其中CTMAB与聚乙二醇-600(PEG-600)合用优于CTMAB或三乙基苄基氯化铵(TEBAC)。3,4,5-三甲氧基扁桃酸在Me3SiCl、NaI、Zn、乙腈作用下经还原反应合成肌松药米库氯铵中间体3,4,5-三甲基苯乙酸,收率91.0%。其中还原反应的较佳原料3,4,5-三甲氧基扁桃酸、Me3SiCl、NaI、Zn的摩尔比为1:1.25:1.23:3[56]。邹永等报道了类似的反应,其中3,4,5-三甲氧基苯甲醛与氯仿、碱和PTC作用制备3,4,5-三甲氧基扁桃酸的最高收率73.3%,3,4,5-三甲氧基扁桃酸钠的还原在二水氯化亚锡、浓盐酸中进行,得到3,4,5-三甲氧基苯乙酸,收率74.4%[57]。
戴康报道4-乙氧基苯甲醛与氯仿、氢氧化钠在TEBAC催化下制得4-乙氧基扁桃酸,收率90%,再以亚磷酸在水/乙醇体系中还原得到4-乙氧基苯乙酸,收率75%[58]。
高莹等报道2,4,6-三甲基苯甲醛与氯仿、氢氧化钠、甲基三辛基氯化铵作用得到α-羟基-2,4,6-三甲基苯乙酸,重结晶后收率52%。再经次磷酸、氢氧化钠还原反应得到杀虫杀螨剂螺虫甲酯中间体2,4,6-三甲基苯乙酸,收率91%[59]。
Milne等报道了1种钯催化的扁桃酸酯脱羟基合成芳乙酸酯的方法。具体工艺是,先以二苯氧基磷酰氯活化扁桃酸酯的羟基,生成磷酸酯型中间体,再经钯催化的硼氢化钠还原脱羟基,其中活化生成磷酸酯的反应以三乙胺为缚酸剂,4-二甲胺基吡啶(DMAP)为催化剂,THF为溶剂。结果表明,还原反应的最佳金属配体组合是Pd(ACN)2Cl2/BINAP(ACN 为乙腈,BINAP 为 1,1′-联萘-2,2′-双二苯膦),最佳还原体系为硼氢化钠、THF、乙二醇二甲醚(DME)。但三乙胺-甲酸、甲酸铵也能得到不错的效果。苯环上带吸电子或给电子的取代基均可获得良好的结果[60]。
芳醛也可以通过与氢氰酸加成得到α-羟基芳乙腈,水解得到扁桃酸,因此经扁桃酸或酯还原脱羟基制备芳乙酸的方法具有较高的实用价值。
2.3.3 经Darzens缩合、水解、氧化反应
芳醛与卤乙酸酯、醇钠等作用,发生Darzens缩合反应生成β-芳基-α,β-环氧丙酸酯,经水解、酸化、脱羧重排得到芳乙醛,氧化得到芳乙酸:

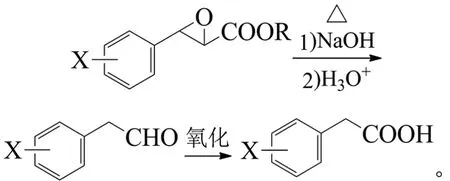
方方等以异香草醛为原料,经苄溴、碳酸钾的苄基化制得3-苄氧基-4-甲氧基苯甲醛,再与氯乙酸乙酯、乙醇钠,经Darzens缩合、氢氧化钠水解、盐酸酸化脱羧重排反应制得3-苄氧基-4-甲氧基苯乙醛,分离浓缩液直接以亚氯酸钠、双氧水氧化制得3-苄氧基-4-甲氧基苯乙酸,以3-苄氧基-4-甲氧基苯甲醛计总收率86%[61]。
李星等报道,3,4,5-三甲氧基苯甲醛与氯乙酸甲酯、碳酸钾、TEBAC、DMF经Darzens缩合、水解、酸化、脱羧重排生成3,4,5-三甲氧基苯乙醛,收率78.4%。再以亚氯酸钠/双氧水氧化得到3,4,5-三甲氧基苯乙酸,收率86.3%[62]。
由以上2例可见,芳醛经Darzens缩合、水解、脱羧重排、氧化制备芳乙酸的方法虽然步骤较多,但多数中间体无需分离,反应条件温和,总收率较高。
2.3.4 其他方法
除以上3种方法外,芳醛还可以通过以下一些特殊方法转化为芳乙酸。
Zhou等报道了1种芳醛与N-(2,3,4,6-四-O-特戊酰化-D-吡喃葡萄糖)胺缩合形成席夫碱,再与三甲硅基腈、溴化亚铜加成得到α-氨基腈,最后在氢溴酸、醋酸、二氯甲烷、水存在下发生碳-氮键断裂,生成芳乙酸的方法:

共研究了10种芳醛,包括羟基、甲氧基、甲基、氯、氟、硝基等取代芳醛,其中生成席夫碱的收率90%~96%,生成α-氨基腈的收率85%~92%,生成芳乙酸的收率为85%~93%[63]。该方法的模板胺N-(2,3,4,6-四-O-特戊酰化-D-吡喃葡萄糖)胺可以套用,反应条件温和,适用范围广,具有一定的实用价值。
Huh等报道,芳醛与四溴化碳、三苯基膦反应生成2,2-二溴芳乙烯,再与胺反应转化为芳乙酰胺,最后经水解得到芳乙酸。第2步转化中,吡咯烷和氢氧化钾为最佳的胺和碱[64]:
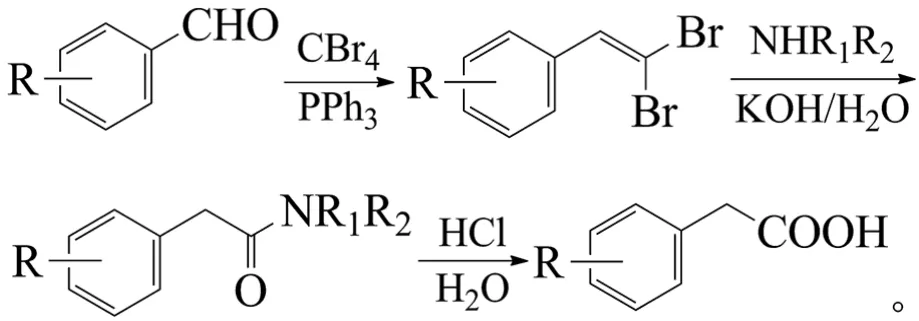
该方法适用范围广,甲基、甲氧基、氯、三氟甲基、氰基、硝基等取代的芳醛均能获得良好收率的芳乙酸(三步反应的收率分别为73%~99%、81%~99%和91%~99%)。该方法试剂易得、条件温和,但四溴化碳和三苯基磷价格较高,分子量大,吡咯烷价格高等,因此主要适合实验室使用。
Williams等报道3-甲氧基-4-乙酰氧基-2-硝基苯甲醛和3-甲氧基-4-乙酰氧基-5-硝基苯甲醛与N-乙酰甘氨酸以醋酸钠/醋酐缩合、盐酸/醋酸水解、双氧水/氢氧化钠氧化三步反应,生成相应的苯乙酸,3步反应总收率达43%[65]。
张黔等报道了类似的合成方法,3,4,5-三甲氧基苯甲醛与N-苯甲酰甘氨酸在醋酸钠、醋酐中缩合得到4-亚苄基-2-苯基恶唑酮,双氧水和氢氧化钠氧化、酸化得到3,4,5-三甲氧基苯乙酸,3步反应总收率58%[66]:

以上芳醛与N-乙酰或N-苯甲酰甘氨酸为原料的3步反应方法,原料价廉易得,但收率偏低,需要进一步改进。
[1]叶盼盼,金耀来,郑土才,等.芳乙酸类化合物的合成研究进展I[J].化工生产与技术,2015,22(1):46-53.
[2]Shabanov A L,Ramazanova E M.Methods of phenylacetic acid production:US2007/010685[P].2007-01-11.
[3]Salman M,Babu S J,Ray P C,et al.An efficient and costeffectivesynthesisof3-ethoxy-4-ethoxycarbonylphenylacetic acid:a key acid synthon of repaglinide[J].Org Proc Res Dev,2002,6:184-186.
[4]仇章明.3-乙氧基-4-乙氧羰基苯乙酸的合成方法:CN 101891621[P].2010-11-24.
[5]钱珊,何宇欣,王周玉.3-乙氧基-4-乙氧羰基苯乙酸的合成研究[J].安徽农业科学,2011,39(29):17907-17908.
[6]Aswathanarayanappa C,Ujire S,Puthiaparampil T T,et al.Ethyl 4-(2-tert-butoxy-2-oxoethyl)-2-ethoxybenzoate,intermediate for the preparation of repaglinide:WO2005/019140[P].2005-03-03.
[7]王迷娟,张秋佳,尹大力.3-乙氧基-4-乙氧羰基苯乙酸的合成[J].中国医药工业杂志,2006,37(6):378-379.
[8]Zheng Z B,Wang A X,Scola P,et.al.Improved synthesis of 1-chloro-6-methoxyisoquinoline-3-ol and its derivatives[J].Synth.Commun,2009,39,1264-1272.
[9]蔡可迎,王桃霞,高媛.邻卤苯乙酸的合成[J].化工进展,2006,25(10):1214-1216.
[10]蔡可迎,丁明洁,宗志敏,等.邻卤苯乙酸合成方法的改进[J].湖北大学学报(自然科学版),2007,29(1):82-84,88.
[11]郭希,杨艺虹,张珩,等.5-(2-氯乙酰基)-6-氯吲哚酮的合成[J].武汉工程大学学报,2010,32(11):25-27.
[12]孙平华,陈卫民,李冰渊.盐酸罗匹尼罗的合成[J].中国医药工业杂志,2007,38(1):7-8.
[13]Qiu Z,He Y,Zheng D,et al.Study on the synthesis of phenylaceticacidbycarbonylationofbenzylchlorideundernormal pressure[J].J Nat Gas Chem,2005,14:40-46.
[14]李剑利,校大伟,万克柔,等.苄基氯羰基化合成苯乙酸钴复合相转移催化体系:CN 101816952[P].2010-09-01.
[15]金洗郎,李剑利,王淑莉,等.苄基氯羰基化合成苯乙酸的方法:CN102050721[P].2011-05-11.
[16]王立中,卞小琴.邻氯苯乙酸的合成研究[J].天津化工,2011,25(1):49-50.
[17]钟建新,何人宝.2,4,5-三氟苯乙酸的制备方法:CN101092345[P].2007-12-26.
[18]何人宝,王莺妹,金逸中,等.2,4,5-三氟苯乙酸的合成[J].化工生产与技术,2011,18(3):4-6.
[19]Jones R V H,Lindsell W E,Palmer D D,et al.Palladiumcatalyzed carbonylation of arylmethyl halides:efficient synthesis of arylacetic acid and esters[J].Tetrahedron Lett.2005,46:8695-8697.
[20]王萍,樊小彬,林行军,等.一种2,5-二甲基苯乙酸的制备方法:CN102140062[P].2011-08-03.
[21]IsseA A,Gennaro A.Electrocatalyticcarboxylation of benzyl chloridesat silver cathodes inacetonitrile[J].ChemCommun,2002,23(23):2798-2799.
[22]陆嘉星,胡磊磊,王欢,等.一种对乙烯基苯乙酸的制备方法:CN101717949[P].2010-06-02.
[23]吴永兰,李广利,文耀智.6-羟基异黄酮的微波合成研究[J].广东化工,2011,38(8):15-16
[24]祝宏,潘盛钢,刘永琼,等.联苯乙酸的合成新工艺研究[J].化学工程与装备,2010(8):18-20,29.
[25]张群辉,黄明旺,陶开跃.三氟苯乙酸和西他列汀的制备方法:CN101429115[P].2009-05-13.
[26]冯秀娟,谭君,杨帆,等.一种2,4,5-三氟苯乙酸的制备方法:CN101823952[P].2010-09-08.
[27]石超君,吕秀阳.近临界水中苯乙腈无催化水解反应动力学[J].高校化学工程学报,2009,23(2):252-257.
[28]吕秀阳,石超君.近临界水介质中苯乙腈无催化水解制备苯乙酸的方法:CN101161626[P].2008-04-16.
[29]傅杰,任浩明,吕秀阳.CO2对高温液态水中苯乙腈水解反应的影响[J].精细化工,2012,29(1):86-90.
[30]任浩明,吕秀阳.含氨高温液态水中苯乙腈水解反应动力学[J].化工学报,2009,60(6):1435-1441.
[31]吕秀阳,任浩明.含氨高温液态水介质中苯乙腈水解同时制备苯乙酰胺和苯乙酸的方法:CN101381325[P].2009-03-11.
[32]吴毅,范谦,程珂.微波辅助合成4-氯-α-羟基苯乙酸[J].化工时刊,2011,25(10):27-28.
[33]Baelen,G.V,Maes,B.U.W.Study of the microwave-assisted hydrolysis of nitriles and esters and the implementation of this system in rapid microwave-assisted Pd-catalyzed amination[J].Tetrahedron,2008,64:5604-5619.
[34]郭新艳,姜申德.番荔枝宁合成工艺[J].精细化工,2010,27(9):882-884.
[35]朱玉松,罗世能,沈永嘉.白藜芦醇类似物的合成[J].有机化学,2006,26(7):958-962.
[36]陈勇,许志献,黄亮.4-(2-噻吩基)苯乙酸及其衍生物的合成[J].化学世界,2006(5):296-298.
[37]Vincze Z,Biro A B,Csekei M,et al.The palladium-catalyzed preparation of condensed tetracyclic heterocycles and their applications to the synthesis of rac-mangochinine[J].Synthesis,2006(8):1375-1385.
[38]陈康,李惠,赵东江.2,4-二氯苯乙酸合成工艺研究[J].精细化工中间体,2010,40(2):16-17.
[39]邹建平,尹笃林,赵东江,等.2,5-二甲基苯乙酸合成工艺改进研究[J].精细化工中间体,2015,45(2):20-22.
[40]李惠,徐西之,赵东江.2,4,6-三甲基苯乙酸的合成研究[J].精细化工中间体,2010,40(1),15-17.
[41]刘宇,赵东江,杨彬,等.2,4,6-三甲基苯乙酸合成工艺改进研究[J].精细化工中间体,2014,44(1):10-12.
[42]宋尚海,金雪光张国鑫.2,4,6-三甲基苯乙腈合成2,4,6-三甲基苯乙酸[J].浙江化工,2010,41(11):5-7.
[43]蔡正艳,宁奇,周伟澄.阿伐麦布的合成[J].中国医药工业杂志,2005,36(8):453-454.
[44]但飞君,田瑛,董俊兴.3,4-二羟基苯乙酸的合成[J].化学试剂,2005,27(10):623-624.
[45]贺新,邱岳进.3,4,5-三甲氧基苯乙酸的合成工艺改进[J].中国现代应用药学,2010,27(1):32-34.
[46]孙晋瑞,刘宜辉,张治法,等.一步法合成4-联苯乙酸[J].食品与药品,2013,15(2):115-117.
[47]Salem O I A,Frotscher M,Scherer C,et al.Novel 5αreductase inhibitors:synthesis,structure-activity studies,and pharmacokinetic profile of phenoxy-benzoylphenyl acetic acids[J].J Med Chem 2006,49:748-759.
[48]Lee J,Lee J H,Kim S Y,et al.2-Benzyl and 2-phenyl-3-hydroxypropyl pivalatesasproteinkinaseCligands[J].Bioorg Med Chem Lett,2006,14:2022-2031.
[49]Nakhostin A,Mirseyyedhosein S,Toolabi M,et al.Syntheis,conforma-tionalassignment,andanti-inflammatoryactivities ofN-arylidene-2-(4-chloro-2-(2-substituted phenoxy)phenyl)acetic acid hydrazides[J].Med Chem Res,2016,25:2220-2236.
[50]李燕,彭光荣,袁云龙,等.2,4,5-三氟苯乙酸的制备方法:CN1749232[P].2006-03-22.
[51]钟建新,邵鸿鸣.2,4,5-三氟苯乙酸的制备方法:CN101659611[P].2010-03-03.
[52]刘一超,王秀云,孙为仁,等.2,4,5-三氟苄氯和2,4,5-三氟苯乙酸的制备方法:CN102690166[P].2012-09-26.
[53]Matt C,Wagner A,Mioskowski C.Novel transformation of primary nitro-alkanes and primary alkyl bromides to the corresponding carboxylic acids[J].J Org Chem,1997,62:234-235.
[54]许慧,蒋玉仁,陈芳军.3,4-二甲氧基苯乙酸的合成新法合成[J].广州化工,2009,37(1):39-41.
[55]Nikalje M D,Ali I S,Dewkar G K,et al.Synthesis of arylαketo-acids via the Cu-catalyzed conversion of aryl nitroaldol products[J].Tetrahedron Lett,2000,41:959-961.
[56]邓旭忠,黄顺,王岳群.3,4,5-三甲氧基苯乙酸的合成[J].精细化工,2011,28(12):1240-1243.
[57]邹永,都建立,陈大峰,等.一种双或多甲氧基取代苯乙酸的制备方法:CN101274888[P].2008-10-01.
[58]戴康.一种制备4-乙氧基苯乙酸的方法:CN102531884[P].2012-07-04.
[59]高莹,聂光辉,方旭兵.2,4,6-三甲基苯乙酸的合成新方法[J].精细化工中间体,2011,41(4):49-51.
[60]Milne J E,Murry J A,King A,et al.Pd-catalyzed deoxygenation of mandelate esters[J].J Org Chem,2009,74:445-447.
[61]方方,吉爱国.3-苄氧基-4-甲氧基苯乙酸的制备[J].中国医药工业杂志,2006,37(2):80-81.
[62]李星,张俊,李杨,等.3,4,5-三甲氧基苯乙酸的合成新工艺研究[J].浙江理工大学学报,2013,30(4):616-619.
[63]Zhou G B,Zhang P F,Pan Y J.A novel method for synthesis of arylacetic acids from aldehydes,N-(2,3,4,6-tetra-O-pivaloylated-D-glucopyranosyl)amine and trimethylsilylcyanide[J].Tetrahedron.2005,61:5671-5677.
[64]Huh D H,Jeong J S,Lee H B,et al.An efficient method for one-carbon elongation of aryl aldehydes via their dibromoalkene derivatives[J].Tetrahedron,2002,58:9925-9932.
[65]Williams R W,Cao J,Tsujishima H,et al.Asymmetric stereocontrolled total synthesisof Paraherquamide A[J].JAm Chem Soc,2003,125:12172-12178.
[66]张黔,张灿.Erlenmeyer-Plochl反应制备3,4,5-三甲氧基苯乙酸[J].药学进展,2013,37(6):280-283.
