Genomic,Lipidomic,and Metabolomic Analysis of Cyclooxygenase-null Cells:Eicosanoid Storm,Cross Talk,and Compensation by COX-1
2016-06-02AbulIslamMandarDaveSoniaAminRoderickJensenAshokAmin
Abul B.M.M.K.Islam,Mandar Dave,Sonia Amin,Roderick V.Jensen,Ashok R.Amin
Genomic,Lipidomic,and Metabolomic Analysis of Cyclooxygenase-null Cells:Eicosanoid Storm,Cross Talk,and Compensation by COX-1
Abul B.M.M.K.Islam1,#,a,Mandar Dave2,3,#,b,Sonia Amin4,c,Roderick V.Jensen4,d,Ashok R.Amin1,2,4,5,6,*,e
1Department of Genomic Engineering and Biotechnology,University of Dhaka 1000,Bangladesh
2Department of Rheumatology,New York University Hospital for Joint Diseases,New York,NY 10003,USA
3Department of Biology and Chewistry,Essex County College,Newark,NJ 07102,USA
4Department of Biological Sciences,College of Sciences,Virginia,Tech Blacksburg,VA 24060,USA
5Department of Pathology,New York University School of Medicine,New York,NY 1006,USA
6RheuMatric Inc.Blacksburg VA 24060,USA
Received 14 October 2015; revised 26 January 2016; accepted 10 March 2016 Available online 21 March 2016
Handled by Xiangdong Fang
KEYWORDS
Metabolomics;
Lung fibroblasts;
Genomics;
Inframmation
Abstract The constitutivly-expressed cyclooxygenase I(COX-1)and the inducible COX-2 are both involved in the conversion of inarachidonic and(AA)to prostaglandins(PGs).However,the functional roles of COX-1 at the cellular level remain unclear.We hypothesized that by camplaing differential gene expression and eicosanoid metabolism in lung fibroblasts from wild type(WT)mice and COX-2-/-or COX-1-/-mice may help address the functional roles of COX-1 in inflammtion and other cellular functions.Campared to WT,the number of specifically-induced transcripts were altered descendingly as follows:COX-2-/->COX-1-/-WT + IL-1β.COX-1-/-or COX-2-/-> cells shared about 50% of the induced transcripts with WT cells treated with IL-1β,respectively.An interactive “anti-inflammatoy,proinflammatory,and redox-activated”signature in the protein interactome map was obseved in COX-2-/-cells.The augmented COX-1 mRNA(in COX-2-/-cells)was associated with the upregulation of mRNAs for glutathione S-transferase(GST),superoxide dismutase(SOD),NAD(P)H dehydrogenase quinone 1(NQO1),aryl hydrocarbon receptor(AhR),peroxiredoxin,phospholipase,prostacyclin synthase,and prostaglandin E synthase,resulting in a significant increase in the levels of PGE2,PGD2,leukotriene B4(LTB4),PGF1α,thromboxane B2(TXB2),and PGF2α. The COX-1 plays a dominant role in shifting AA toward the LTB4pathway and anti-inflammatory activities. Compared to WT,the upregulated COX-1 mRNA in COX-2-/-cells generated an“eicosanoid storm”. The genomic characteristics of COX-2-/-is similar to that of proinflammatory cells as observed in IL-1β induced WT cells. COX-1-/-and COX-2-/-cells exhibited compensation of various eicosanoids at the genomic and metabolic levels.
E-mail:ashokamin2004@yahoo.com (Amin AR).
#Equal contribution.
aORCID: 0000-0002-7274-0855.
bORCID: 0000-0001-9996-1159.
cORCID: 0000-0002-5751-6169.
dORCID: 0000-0003-3259-0140.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2014.09.005.
1672-0229Ⓒ2016 The Authors. Production and hosting by Elsevier B.V. on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
Introduction
Eicosanoids are lipid mediators of inflammation[1]. Cytosolic phospholipase A2(cPLA2)cleaves some membrane lipids to generate 20-carbon arachidonic acid(AA),which can be channeled toward eicosanoid synthesis[1]. The generation of AA by cPLA2is a rate-limiting step for biosynthesis of eicosanoids,which include prostaglandins(PGs),thromboxanes(TXs),and leukotrienes(LTs). PGs and TXs,which are collectively referred as prostanoids,are generated by the two isoforms of cyclooxygenases(COX),COX-1 and COX-2,whereas LTs are generated by 5-lipoxygenase(5-LOX)[1,2]. Although both COX enzymes generate PGs,COX-1 is constitutively expressed in most normal cells at the basal level and is considered to regulate a number of housekeeping functions such as vascular hemostasis,renal blood flow,and glomerular function [1,2]. On the other hand,COX-2,which is inducible in nature,is mostly expressed at sites of inflammation and cancer[1,2]. Expression of COX-2 can be triggered by cytokines,growth factors,and other proinflammatory stimuli[2]. Cell- and/or tissue-specific gene expression of COX-1 and COX-2 has been reported in different organs and tumors[3-5]. Moreover,COX-2 overexpression induces tumors in COX-2 transgenic mice,whereas the COX-2 inhibitors coxibs induce oxidative stress and reduce polyps associated with colon cancer[2,6,7].
Fibroblasts have a wide distribution throughout the body. Owing to their ability to secrete extracellular matrix(e.g.,collagen)and release proinflammatory mediators[8],fibroblasts are involved in various pathophysiological conditions such as arthritis,cancer,lung fibrosis,wound healing,and stem cell maturation[8]. Fibroblastic COX-1 and COX-2 activity[8]can be modulated by various receptors such as PGE receptors (EP receptors)and Toll-like receptors(TLRs)[1,2,8]. These receptors may influence eicosanoid synthesis,growth,chemotaxis,matrix,and matrix metalloprotease synthesis[1,2]. We therefore chose to use fibroblasts over macrophages for the present study,in line with our previous investigations on the regulation of COX and PGs in peripheral blood leukocytes,chondrocytes(differentiated fibroblasts),and synovial fibroblasts in human arthritis[9-11]. We expect our systems approach(genomic,metabolomic,protein-interatomic,and proteomic)from the same cells will allow identification of common and distinct functions of the two closely related COX isozymes[12,13]in sterile inflammation,innate immune response,and homeostasis in the cells.
The uncontrolled production or defective expression of COX-1 or COX-2 has been recognized as a health risk [14,15]. Similarly,nonsteroidal anti-inflammatory drugs (NSAIDs)and COX-2 inhibitors(coxibs)also exhibited health risk in large-scale population studies[16,17]. The Food and Drug Administration(FDA)of the United States has recommended that coxibs be avoided in individuals with an elevated risk of cardiovascular disease(CVD)and in patients with established CVD[16,17],since the drugs increased the risk of ischemic CVD,heart failure,increased blood pressure,and cardiac arrhythmia[2,16,17]. One explanation of these adverse events is the differential accumulation of TXs,LTs,and PGs in the presence of coxibs and/or decreased level of prostacyclins [2,6-8,9-11]and/or“non-PG effects”of COX inhibitors [18-20]. Inhibition of COX-2 by coxibs in human arthritis not only curbs COX-2-mediated PGs but also shifts prostanoid synthesis toward the COX-1-mediated pathway[3,10]. Unlike COX-2,the participation of COX-1 has been subverted,for its involvement in inflammation[1,2]. For example,the anti-inflammatory effects of a highly-selective coxib were evident only when administered at doses that are inhibitory for COX-1,indicating a substantial contribution of COX-1 in inflammation[2].
IL-1β is a proinflammatory factor with a wide spectrum of metabolic,physiological,homeostatic,inflammatory,sterile inflammatory,and immune activities in diseases[21-23]. Sterile inflammation is caused by cell damage in trauma,ischemia,ischemia-reperfusion,etc. via endogenous ligands,such as high-mobility group protein B1(HMGB1),amyloid,S100 proteins,and heat shock proteins(HSPs)[24,25]. It has been implicated in complex diseases,with the upregulation of IL-1 and eicosanoids as the common denominator[1,9,21-23].
In the present study,we report the alternation in gene expression and end products of lipid synthesis for COX-1,COX-2,and 5-LOX pathways in COX-1-/-(COX-1-ablated)and COX-2-/-(COX-2-ablated)cells. We demonstrate the dominant role of COX-1 at the genomic,redox,and metabolomic levels in COX-2-null cells. In addition,gene array data also agree with the metabolic activity of eicosanoids and redox changes in COX-2-/-cells.
Results
Gene expression arrays and hierarchical clustering in mouse fibroblasts
In the present study,we utilized knockout cells to eliminate the variables previously observed when using COX-1/COX-2 inhibitors to dissect the functional roles of COX-1/COX-2,such as“non-PG effects”,specificity- and concentration-dependent outcomes[3,12,13,16-20].
Mouse fibroblast cells were obtained from WT cells(WT),COX-1-/-,and COX-2-/-mice. In addition,WT cells stimulated with 10 ng/ml IL-1β served as a positive control to procure an“IL-1β inflammatory signature”at the genomic level. All the cells were subjected to gene expression arrays[26-28]and as shown in Figure 1A. We implemented bioinformatics analysisto identify specific transcripts that were upregulated by 1.75-fold or downregulated by 0.5-fold in COX-1-/-,COX-2-/-,and IL-1β stimulated WT cells,respectively,when compared to basal gene expression in WT cells. Hierarchical clustering of gene transcripts showed altered gene expression in COX-1-/-and COX-2-/-cells as compared to WT cells(Figure 1B;Figure S1 for high resolution and gene annotations). These also include COX-1 and genes involved in eicosanoid synthesis,inflammation,homeostasis,and cell cycle in COX-2-/-cells.
As a result,we found expression of 223 transcripts was upor downregulated in IL-1β-induced WT cells(WT + IL-1β),representing the“IL-1β inflammatory signature”(Figure 1C,Figure S2,and Table S1). About 50%of these 223 transcripts were also modulated as the IL-1β inflammatory signature in COX-1 or COX-2 ablated cells(Figure 1C,S2 and Table S1). Expression of some highly or lowly modulated transcripts by IL-1β[21-25],such as acute phase protein serum amyloid A3(SAA3)and IL33 was further plotted(Figure S3). As expected,IL-1β induced the expression of COX-2 but not COX-1,comparing WT vs. WT + IL-1β cells[21-25].

Figure 1 General scheme for experimental conditions,bioinformatics analysis,and hierarchical clustering of genes
Gene expression arrays of eicosanoid metabolism
To understand the role of COX-1,we focused on the alterations in gene expression in COX-2-/-cells. Sixty-five transcripts were identified to be involved in eicosanoid,lipid,and redox metabolism pathways. These transcripts were grouped into different eicosanoid pathways as shown in the heat map of differentially-expressed transcripts from WT,WT + IL-1β,COX-1-/-,and COX-2-/-cells(Figure 2). More than 25% of these 65 transcripts that were upregulated in IL-1β stimulated WT cells were also spontaneously augmented in COX-2-/-cells.
The genes of PLA2-group[29],including Pla2g4,Pla2g6,and Pla2g7,were highly expressed in COX-2-/-followed by COX-1-/-,IL-1β+ WT,and WT cells(Figure 2 and Table S2). Compared to WT and COX-1-/-cells,increased expression of genes encoding PGE synthase(Ptges)and PGI2(as called prostacyclin)synthase(Ptgis)was detected in COX-2-/-cells. Glutathione S-transferases(GSTs)arekey enzymes in the synthesis of various LTs[30,31]. There was an increase in genes encoding various GST isoforms,including GST theta 1(Gstt1),GST alpha 4(Gsta4),GST kappa 1(Gstk1),and microsomal GST 1 and 2(Mgst1, Mgst2),in COX-2-/-cells as compared to WT,WT + IL-1β,and COX-1-/-cells. Similarly,increased expression was also revealed for genes encoding superoxide dismutase (Sod),peroxiredoxin(Prdx),and proteins involved in the phospholipid translocation,such as scramblase 2(Plscr2)[30-33].

Figure 2 Gene expression of eicosanoid metabolism in COX-1-/-,COX-2-/-,WT,and WT + IL-1β cells
Network analysis
The Database Search Tool for the Retrieval of Interacting Genes/Proteins(STRING)contains information on about 5.2 million proteins in 1133 species from experimental data,computational prediction methods,and public text collections. Data are weighted and integrated with a confidence score calculated for all protein interactions. STRING allows merging of gene expression arrays and metabolic activity at the level of protein-protein interactome[13,34]. Using the edge information present in the STRING database,protein-protein interaction map(Figure 3)was derived from the gene expression data in this study. In WT cells,there exist interactions of different proteins in the eicosanoid pathways(Figure 3A),representing the background signal of WT cells. The map shows the interactions among the functional genes/proteins associated with 5-,12-,and 15-LOXs,COX-1 and COX-2,and lipid metabolism. Figure 3 showed an increase in gene/ protein activities in the following order of magnitude: COX-2-/->COX-1-/-= IL-1β+ WT>WT cells. The differences in redox-related protein were more pronounced in COX-2-/-cells(Figure 3D). For example,expression of NAD(P)H dehydrogenase quinone 1(NQO1)and its receptor aryl hydrocarbon receptor(AhR)was increased in COX-2-/-as compared to WT,COX-1-/-,and IL-1β-stimulated cells. NQO1 is induced during cellular stress,detoxification,tumors,tardive dyskinesia,and hematotoxicity[35,36]. AhR has been shown to regulate xenobiotic-metabolizing enzymes such as cytochrome P450,which is involved in eicosanoid metabolism[36]. Taken together,these interaction maps showed that unlike WT and COX-1-/-cells,COX-2-/-cells exhibited a distinct proteinprotein interaction signature.
Lipidomic studies and strategy
Accumulation of PGE2and PLA2in COX-1-/-and COX-2-/-cells
We integrated the omics analysis as shown in Figure S4[37]. We next selected stable end-metabolites(from WT and COX mutants),which can be quantitatively assayed and may also represent pathways involved in eicosanoid synthesis. The expression levels of genes encoding isoforms of PLA2of the groups VII(Pla2g7),IVA(Pla2g4),and VI(Pla2g6)were spontaneously upregulated in the following order: COX-2-/->COX-1-/-= IL-1β+ WT cells(Figure 2 and Table S2). Basal levels cPLA2were not detected by western blot analysis in unstimulated fibroblasts in this and other studies[3,29]. Nevertheless,COX-1-/-and COX-2-/-cells(and WT + IL-1β cells[3,38])showed upregulation of cPLA2as determined by Western blot analysis(Figure 4). These data not only confirmed previous observations[29]with respect to the functional role of cPLA2,but also identified new classes of PLA2: the lipoprotein-associated phospholipase A2(Lp-PLA2)encoded by Pla2g7,which is associated with eicosanoid metabolism in these mutant fibroblasts.
Consistent with the upregulated expression of cPLA2in COX-ablated cells,the levels of PGE2were significantly increased(3-4-folds)in COX-1-/-and COX-2-/-cells,although it is present at a basal level in WT cells(Figure 4A). Similar observations were made after normalizing the amount of PGE2with the cellular DNA content(Figure 4B). We obtained the similar results as those reported by Ballou et al.[3]. The increased levels of PGE2was accompanied by increases in the expression of Ptgs1,Ptgs2,Acsl4,Elovl6,Fads1,Plscr1,Plscr2,Ptgis,and Ptges,which are involved in lipid metabolism(Figure 2 and Table S2). These results suggest an increase in the synthesis and compensation of the dominant PG(e.g.,PGE2)with other enzymes in lipid metabolism by COX-1 and COX-2 pathways in the absence of either COX isoforms.
Regulation of PGE2in COX-1- and COX-2-ablated cells
We then examined if the expression of COX-1 and COX-2 was sensitive to inhibitors(indomethacin)and activators(IL-1β and/or AA)of COX-1 and COX-2 in WT,COX-1-/-,and COX-2-/-cells(Table 1). We found that indomethacin significantly inhibited PGE2accumulation in COX-1 or COX-2-ablated cells,confirming that both enzymes were independently involved in the generation of PGE2[1,2]. On the other hand,we also tested the effects of the endogenous activator AA(Figure 5)and found that AA significantly increased the levels of PGE2in WT and COX-1-/-cells,but not in COX-2-/-cells. These experiments show the importance of endogenous AA as a rate limiting step for regulation of COX-1 and COX-2 pathways[1-3,39].
Analysis of prostanoids in COX-1-/-and COX-2-/-cells
PGE2is one of the dominant prostanoids in COX-ablated fibroblasts as observed in other cell types[1,2]. We examined the production of PGD2,TXB2,6-keto-PGF1α,and PGF2α,which has not been examined previously in these COX mutants(Figure 6). The amount of PGD2was significantly increased in the COX-1-/-and COX-2-/-cells as compared to that in WT cells. Compared to WT cells(around 170-180 pg/ml),the amount of TXB2,a stable and an inactive metabolite of TXA2,was significantly increased in COX-2-/-but not COX-1-/-cells. Different from PGD2and TXB2,PGF1αwas identified in low concentrations in WT cells,but increased significantly in both COX-1-/-and COX-2-/-cells. On the other hand,PGF2αlevels were relatively high in WT cells but significantly increased in COX-2-/-cells,while only marginal increase was detected in COX-1-/-cells. In addition,there was a 1-5-fold increase in several prostanoids(like PGE2)in the presence of exogenous AA(data not shown). In summary,as compared to WT cells,the levels of TXB2and PGF2αwere significantly higher in COX-2-/-cells but not COX-1-/-cells. There was a compensation of basal levels of PGE2,PGD2,and PGF1αin COX-1-/-and COX-2-/-cells due to increased activity of cPLA2and high levels of common substrate: AA in both mutants. This leads to an imbalance in the basal levels of prostanoids in the mutants(by the COX-1 and COX-2 pathways)as compared to WT cells.
Cross-talk between COX and 5-LOX pathways in COX-2-/-cells The shift from PGE2to LTB4in cytokine-activated cells in the presence of coxibs has been reported in human arthritisaffected tissues and cells[9,40]. In the present study,we compared the contributions of COX-1 and COX-2 in the 5-LOXpathways with and without any stimulation(Figure 7). COX-2-/-and COX-1-/-cells showed a spontaneous release of LTB4,although it was much higher in COX-2-/-cells. In addition,extra AA or IL-1β significantly increased the levels of LTB4accumulation in COX-1-/-cells,which is not the case for COX-2-/-cells. In summary,we demonstrated the differential and preferential contribution of LTB4 synthesis by the COX-1 pathway over the COX-2 pathway.
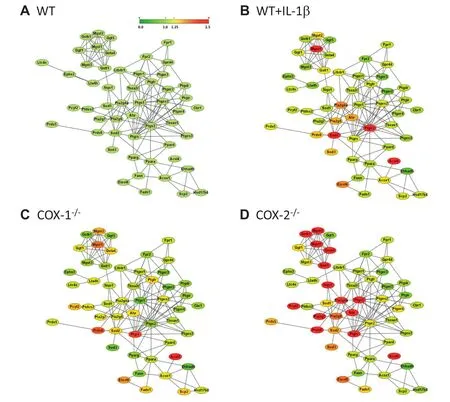
Figure 3 Protein-protein interactions in eicosanoid metabolism
Discussion
The present study describes the common and dissimilar functional interactions of the COX isozymes at the cellular level. Previous pharmacogenomics analysis showed inhibition of PGE2by coxib in human arthritis-affected blood cells and cartilage,resulted in altered gene expression[9-11]. Coxibs or statins with different chemical structures and similar pharmacological targets,elicited distinct side effects,changes in protein profile and gene expression[1,2,16-19,41-43]. It should be noted that altered gene expression incited by coxibs may not necessarily account for the inhibition of COXs and PGs,since some effects of coxib treatment cannot be reversed by the addition of exogenous prostanoids[18-20,42,43]. Furthermore,the differential physiological actions of coxibs for curbing pain and inhibiting colon cancer are concentration dependent[1,2].
Role of COX-1 and COX-2 in inflammation and inflammation resolution
IL-1β and other members of the growing superfamily of IL-1 are involved in sterile inflammation under various pathophysiological conditions[21-25]. Approximately 50%of transcripts modulated in COX-1- or COX-2-ablated cells were also observed in the“IL-1β inflammatory signature”. Since inflammasome and COX-2 pathways are induced by IL-1β,theintricate roles of COX-2 during sterile inflammation and innate immunity were not surprising[23,24]. However,the inflammatory response exhibited by the upregulated COX-1 expression(in the absence of COX-2 activity)was certainly perplexing. These results further support the previously overlooked role of COX-1 within and beyond eicosanoid metabolism and inflammation. Others and we have shown that low levels of PGE2exhibit proinflammatory activity by activating NFκB pathway[44]. However,prolonged activation of COX by IL-1β and high levels of PGE2foster inflammation resolution by inhibiting the NFκB pathway[45,46]. Prolonged and a constitutive upregulation of PGE2(in COX-ablated cells and IL-1β-stimulated WT)triggered the upregulation(by 2-3-fold)of a gene(Nfkbiz),which inhibits NFκB functions[44].
COX-1 or COX-2 in collaboration with other mediators may participate in inflammation,inflammation resolution,and cancer[1,2,45,46]. The multifunctional transcription factor AhR regulates many genes including COX-2,but not COX-1[36,47-52]. AhR-/-mice have been reported to develop heightened inflammatory responses with decreased induction of COX-2,lipid peroxidation,and oxidative stress[52]. AhR agonist leflunomide,which is used for the treatment of rheumatoid arthritis(RA),exhibits anti-inflammatory activity [51]. However,another agonist of AhR,TCDD,induces COX-2(but not COX-1)transcription in cancer due to the presence of the AhR binding site(xenobiotic response elements,XRE)in the COX-2 promoter region[36,47]. The upregulation of AhR was only observed in COX-2-/-but not in COX-1-/-cells. The COX-2-/-cells(with amplified COX-1 activity)also exhibited increased oxidative stress with elevated levels of NQO1,GSTs,PRDX2,and SODs. The distended levels of AhR in COX-2-/-cells may participate in not only detoxification [35,36],but also anti-inflammatory activity[51]. These preliminary observations require further experimental studies to better understand the functional relationships between AhR and COX-1.
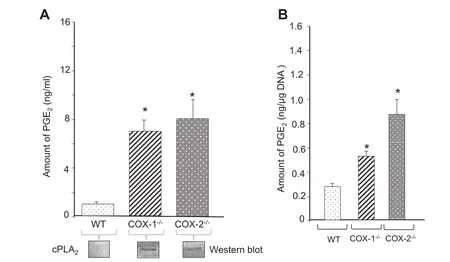
Figure 4 Spontaneous production of PGE2

Table 1 Effect of indomethacin on PGE2production in COX-1- and COX-2-ablated cells
Role of PLA2isoforms and COX-1 in inflammation
In the current studies,the differential expression of various PLA2isoforms in COX-1-/-and COX-2-/-cells were induced by the exclusive COX-2 or COX-1 pathways,respectively.The current literature strongly implicates PLA2G4 as the primary enzyme in polyunsaturated fatty acid release for eicosanoid biosynthesis[12,29]. Pla2g4-/-mice were not able to produce eicosanoids[29,37]. The upregulated Pla2g4 in IL-1β-induced WT,COX-1-/-,and COX-2-/-cells accounted for the increased eicosanoid synthesis[3,29,37]. Transfection of Pla2g6 results in upregulation of PGE2and PGF2α[29,37]. Indeed,unlike in COX-1-/-cells,an increase in Pla2g6 was associated with increased levels of PGF2α.in COX-2-/-cells in the present study. Lp-PLA2(Pla2g7)was upregulated in COX-2-/-cells and plaque forming inflammatory cells in atherosclerosis[29,37]. The differential expression of isoforms of PLA2supports the distinct role of COX-1 in mounting an inflammatory response in the absence of COX-2.
Our studies suggest that COX-1 upregulates gene expression and metabolic activity,which results in a significant increase in the synthesis of prostanoids and redox-related activity in COX-2-/-cells. It should be noted that COX-2-/-fibroblasts showed a nearly fourfold increase in COX-1 gene expression as compared to WT cells. Although COX-1 is regarded as a“constitutive enzyme”[1-3],COX-1 gene expression is upregulated in the colon and ovarian cancers[53,54]. Estradiol also stimulates gene expression of PLA2and COX-1 in endothelial cells[55]. Von Moltke et al. studied the role of COX-1 during systematic inflammation induced by flagellin in mice[56]. They showed that COX-1-derived products drive the initial phase of the inflammatory process,whereas COX-2 upregulation followed a few hours later[56]. These observations highlight the new role of COX-1-generated eicosanoids and physiological consequences in vivo and also support the hypothesis that the basal activity of COX-1,when placed in a pivotal position,can mount an inflammatory response with an“eicosanoid storm”to compensate for the multifunctional eicosanoids,lipids,and redox-related mediators.
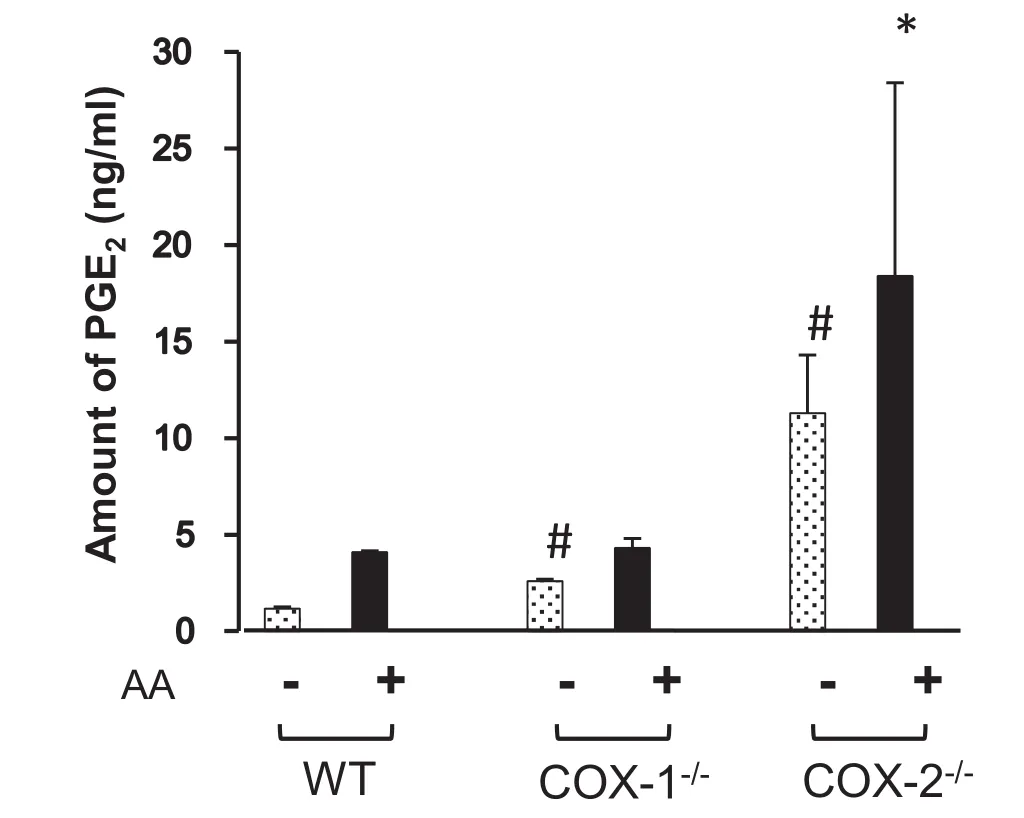
Figure 5 Regulation of PGE2in the presence or absence of AA
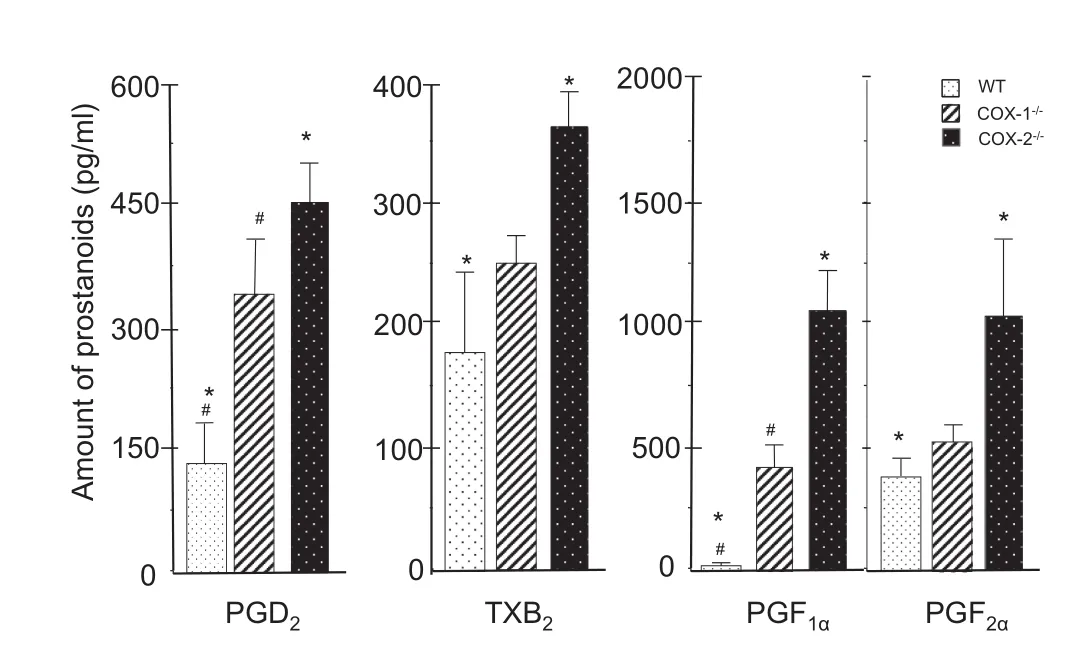
Figure 6 Differential synthesis of prostanoids in WT,COX-1-/-,and COX-2-/-cells
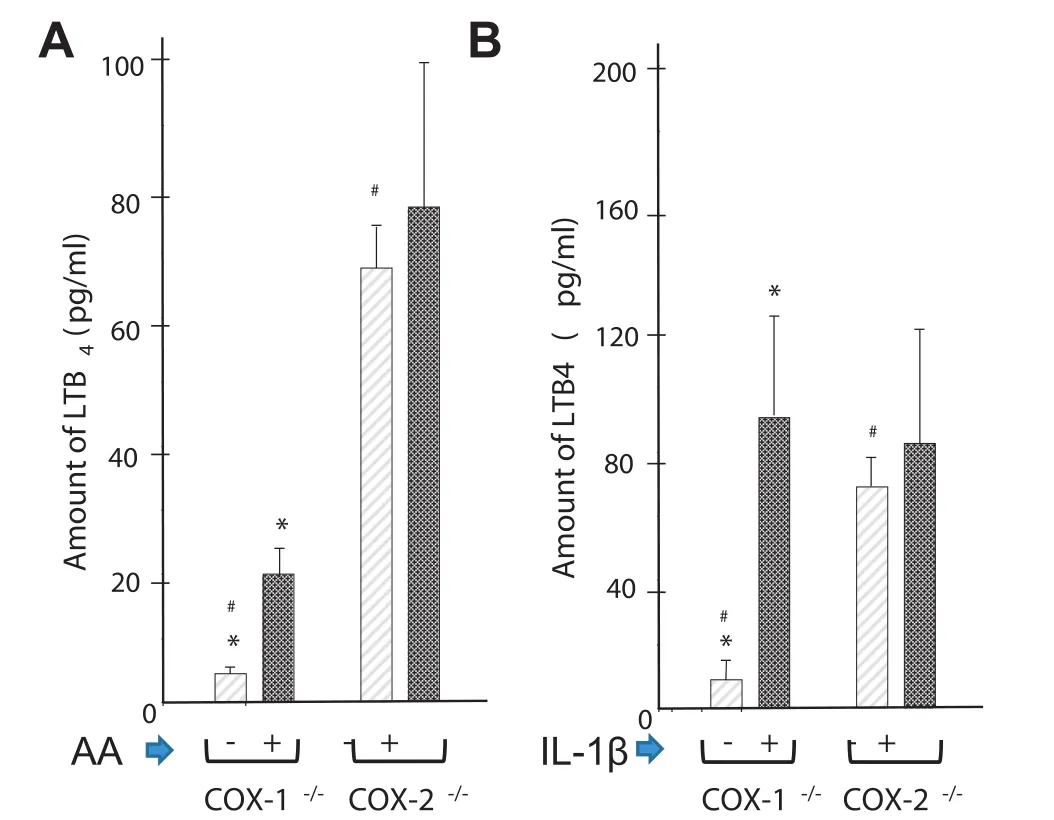
Figure 7 Effect of AA and IL-1β on the level of LTB4in COX-1-/-and COX-2-/-cells
Regulation of metabolites by COX-1,COX-2,and LOX
COX-1 and COX-2 differ in metabolic functions. Unlike COX-1,COX-2 can metabolize dihomo-γ-linolenic andeicosapentaenoic acid in addition to AA[37]. Another noteworthy difference between COX-1-/-and COX-2-/-fibroblasts is their ability to synthesize different quantities and forms of PGs,TXs,LTs,and lipids. Changes in levels of eicosanoids may regulate each other. For example,an increased production of PGD2in human arthritis-affected tissues increases the accumulation of PGE2,PGF1α,PGF2α,and TXB2but inhibits LTB4[57]. The expression of COX-1 can have far-reaching effects on lipid metabolism. For example,Ma et al. reported significant changes in the levels of phosphatidylserine,triacylglycerol,and cholesterol,which alter the cholesterol-tophospholipid ratio in COX-2-/-mice and may impart resistance to neuroinflammation[58]. Thus increasing the activity of COX-1(in COX-2-/-cells)not only regulates eicosanoid synthesis but also augments lipid metabolism as described in Figure 2. An increased activity of COX-1 increased the levels of LTB4in COX-2-/-cells that were comparable to those induced by IL-1β- or AA-induced COX-1-/-cells. Similar effects were observed in human osteoarthritis-affected cartilage that spontaneously released PGE2and LTB4in ex vivo models[9]. The proinflammatory activity of LTB4is well documented in fibroblasts. Expression of TNFα and IL-1β is increased by LTB4but inhibited by LTB4inhibitors,such as MK886 and bestatin[1,2].
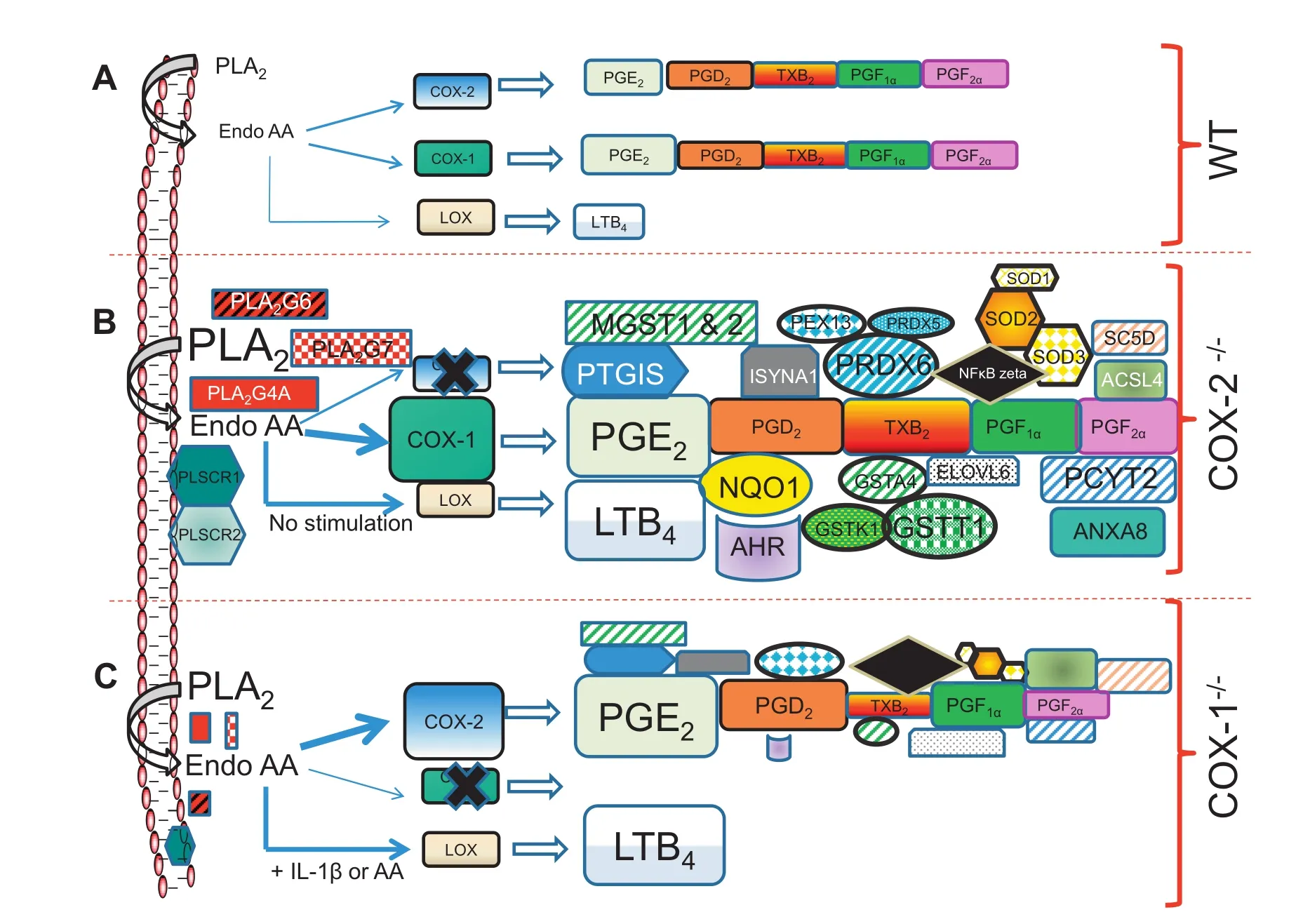
Figure 8 Differential regulation of COX-1 and COX-2- in fibroblasts
Increase in redox molecules in COX-ablated cells
The increase in GSTs in COX-2-/-cells was higher than that in COX-1-/-cells,followed bythat in WT cells. GSTs contribute to the detoxification of xenobiotics and endogenous peroxided lipids,and biosynthesis of LTs[30-32]. Peroxiredoxins function as antioxidant enzymes that also control inflammation-induced peroxide levels by removing H2O2[59]. Mice lacking peroxiredoxin develop severe hemolytic anemia and hematopoietic cancers,and have shortened lifespan[59]. Microsomal PGE synthase-1(mPGES-1)requires glutathione as a co-factor [37]. One of the most notable observations is the increase in gene expression of NQO1 and AHR. AhR-mediated NQO1 gene expression is increased by a variety of antioxidants,tumor promoters,and H2O2[35,36]. COX-2-/-cells showed an increase in the pentose phosphate pathway(HMP shunt,Amin AR unpublished data). The HMP shunt is a metabolic redox sensor and modulates gene expression during an anti-oxidant response [60]. These types of cellular events that lead to oxidative stressand“Warburg effect”[60]are interlinked with the regulation of COXs,NFκB activation during inflammation induces intracellular ROS[46,47,60]. COX-1 peroxidase activity has been reported to serve as an intracellular signal leading to NFκB activation[47,60]. The possibility that the products of COX-1 and COX-2 may also be sensors and/or inducers for such imbalanced redox activity is compelling.
The current understanding of COX-1 in inflammation and homeostasis is constantly evolving. These studies advocate that COX-1 may participate in regulating sterile inflammation,which is more latent and subtle. There are significant collaborations and functional redundancies between COX-1 and COX-2. For example,the essential and collaborative role of both COX-1 and COX-2 has been observed in ear inflammation and arthritis models of mice[1,61]. In addition,in the K/BxN serum-transfer model of arthritis,COX-1-derived PGs,in particular PG12,contributed remarkably to initiating and prolonging the pathology[1]. Similarly,both COX isoforms contributed to PG production in the carrageenan model of inflammation,depending on the type of stimulus and tissuespecific expression of COXs[1]. The coordination and collaboration of the COXs remain well balanced,especially in vivo,in an inflammatory process,but may depend upon the availability ofthe COX isoform,timecourse,andintensityoftheinflammation. It is apparent that if either COX isoform is functionally unavailable,the other becomes pivotal and attempts to compensate the events with a different intensity and/or tissue specificity[9],as observed in COX-1 and COX-2 knockout mice and fibroblasts[3-10,14,15,39]. Figure 8 summarizes the changes observed in WT,COX-2-/-,COX-1-/-cells based on genomics and metabolomics studies described in this study These investigations paint a broader picture of eicosanoid synthesis and signaling,gene expression,and lipid metabolomics,which can identify and connect unforeseen functions of COX-1 and COX-2.
Conclusion
This study describes the functional role of COX-1 and COX-2 at the cellular,metabolic,and genomic levels. Dysregulation of COX-1 or COX-2 results in a burst of gene expression within and beyond eicosanoid and redox metabolism in fibroblasts. The changes in basal gene expression of upregulated COX-1 in COX-2-/-cells exhibited a mixture of pro- and antiinflammatory response,as well as an activated redox signature at the genomic level. The basal levels of COX-1>COX-2 are involved in channeling LTs from COX to 5-LOX pathway. There is compensation of eicosanoids and redox metabolism by COX-1 and COX-2 and vice versa. This systems approach allows the integration of genomic,bioinformatic,and lipidomic datasets. It opens new areas of biomarkers and COX regulation beyond eicosanoid metabolism,which would help to develop safer next generation of coxibs by taking into consideration the board impact of COX genes on other metabolic pathways.
Materials and methods
Reagents
Cell culture media and fetal calf serum(FCS)were obtained from Gibco BRL(Invitrogen,Carlsbad,CA). ELISA kits for the detection of PGs(PGD2,PGF1α,and PGF2α),TXs (TXA2),and LTs(LTB4)and AA were purchased from Chemicon International(Temecula,CA). ELISA was used to measure the levels of prostanoids and LTB4[8,9]. PGE2was estimated by radioimmunoassay(RIA)as previously reported [9,62,63]. IL-1β was purchased from Pepto Tech(Princeton,NJ).
Isolation and culture of COX-deficient mouse cells
Lung fibroblast cells were collected from WT C57BL/6J(B6),as well as COX-1 and COX-2 deficient mice[3-5]. The COX-1-/-and COX-2-/-cell lines were immortalized and cultivated from fibroblasts as previously reported[3-5,62]. Briefly lung tissues were dissected into small pieces and grown in MEM medium[3-5,62],with 10%FCS in a humidified incubator with 5%CO2. After 3 weeks of culture,only fibroblasts continued to grow. The cells were maintained in DMEM containing high glucose and supplemented with Pen/Strep(100,000 U/l penicillin G and 100 mg/l streptomycin sulfate),nonessential amino acids(0.1 mM),fungizone(1 mg/l amphotericin B),glutamine(292 mg/l),ascorbic acid(50 mg/l),and 10%FCS. The cells were incubated with 0.5 μM of AA or 10 ng/ml of IL-1β as endogenous or receptor mediator inducer of eicosanoids.
Labeling and hybridization of microarray gene chips
Total RNA was isolated and labeled with the ENZO BioArray High-yield RNA transcript labeling kit(Affymetrix,Santa Clara,CA)and then hybridized using U74Av2 Murine Genome Array(Affymetrix)as described[26]. Genes representing 12,492 transcripts from the A chip and those annotated till 2012 were identified as present calls as per the manufacturers’recommendations.
Normalization of the microarray data and bioinformatics analysis
The Affymetrix microarray image files for each sample were imported into the Affymetrix Expression Console Software package(www.affymetrix.com)to calculate the robust multiarray analysis-normalized gene expression signals for 12,488 mouse gene probe sets on the U74Av2 arrays. The binary logarithm signal values were then exported into a MS Excel spreadsheet for data analysis. The log transformed expression values ranged from 6.2 to 15.4. Since the median standard deviation of the logged signal values across all probe sets for the replicate samples was small(<0.03),the fold change (FC)of the gene expression was computed by simply averaging the logged signal values for the replicate samples for comparison with the logged expression values for a single WT control sample. Our functional genomics analysis focused primarily on genes with FC≥1.75-fold(upregulation)and≤0.5-fold (downregulation),in relation to WT[26-28]. The normalization was verified using four housekeeping genes encoding glyceraldehyde-3-phosphate dehydrogenase(GAPDH)and β actin,as well as ribosomal proteins L30(RPL30)and S13 (RPS13),which showed similar expression in all samples[64]. The heatmaps were constructed using Gitools[65]with gene expression in the WT taken as the baseline for each transcript.
Hierarchical clustering
The hierarchical clustering of median expression value for every gene was calculated and subtracted from the absolute expression of each gene in each aforementioned experimental condition(median centering of genes). Using the tool“Genesis”and the median-centered gene expression matrix,the hierarchical cluster of genes(Pearson’s correlation distance and average linkage)was determined and the annotated data were presented as heatmaps.
Interaction network
Interactions among proteins encoded by the selected group of eicosanoid metabolism genes were presented in a network model. Edge information among the nodes was extracted using the online tool STRING(v9.1)[66]and the interaction network was visualized using Cytoscape(v 3.1)[67].
Eicosanoids detection in WT,COX-1-/-,and COX-2-/-cell
supernatants
Supernatants of WT,COX-1-/-and COX-2-/-cells were analyzed using ELISA or RIA[8,9]to quantitate eicosanoids. The cells were seeded in triplicates unless otherwise specified and cultured for 24 h.
Effects of AA on eicosanoid synthesis in COX-ablated cells
Cells were seeded in triplicate at a concentration of 10,000 cells/cm2in 6- or 24-well plates. After treatment with 0.5 μM AA for 15 min,the cells were washed and replenished with fresh medium. The levels of eicosanoids were monitored since then and the experiments were terminated between 18 and 24 h before the fibroblasts start the exponential growth. Release of eicosanoids into the medium was subsequently estimated using ELISA or RIA as previously reported[8,9,63].
Western blot analysis
The WT,COX-1-/-,and COX-2-/-cells were cultured for 18 h,and cell extracts were prepared using a total protein extraction reagent(Pierce,Rockford,IL)as per manufacturer’s instructions and other studies[3,62,63]. Total protein(30 μg per sample)was separated by 10%SDS-PAGE and transferred onto a nitrocellulose membrane(Schleicher & Schuell,Keene,NH). After blocking with 3%BSA for 2 h,the blots were incubated with rabbit anti-cytosolic-PLA2antibody(1:500,Santa Cruz Biotechnology,Santa Cruz,CA). Bound antibody was detected by anti-rabbit IgG-conjugated horseradish peroxidase (1:5000,Santa Cruz Biotechnology,Santa Cruz,CA)and developed using the enhanced chemiluminescence ECL plus (Amersham,Arlington Heights,IL).
Statistical analysis
The statistical analyses were performed using GraphPad Software(v1.14)(San Diego,CA). Student’s t-test was used to analyze the data unless otherwise specified. Data from each experiment are represented as mean±standard deviation. Differences between the mean values of the control and experimental groups are considered significant with P<0.05.
Authors’contributions
ARA conceived the project and designed the experiments. ABI and RVJ were involved in interpretation of gene expression data and bioinformatics. SA annotated data and prepared the figures. MD performed eicosanoid assays. All authors participated in the preparation of the manuscript,read and approved the final manuscript.
Competing interests
The project was partially supported by a Translational Research and Target Discovery Contract in collaboration with Yamanuchi Pharmaceuticals(Astellas).
Acknowledgments
We would like to thank Dr. Leslie. Ballou,Department of Veterans Affairs Medical Center,Memphis,Tennessee for providing the lung fibroblasts,COX-1-/-and COX-2-/-cells used for this study. Dr. Mary Leung performed some of the PG assays for which we are very grateful. This project was supported in part by NIH(Grant No. AR 47206-03),USA and Yamanuchi Pharmaceuticals,Japan.
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2014.09.005.
References
[1]Ricciotti E,FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31:986-1000.
[2]Williams CS,Mann M,DuBois RN. The role of cyclooxygenases ininflammation,cancer,anddevelopment. Oncogene 1999;18:7908-16.
[3]Kirtikara K,Morham SG,Raghow R,Laulederkind SJ,Kanekura T,Goorha S,et al. Compensatory prostaglandin E2 biosynthesis in cyclooxygenase 1 or 2 null cells. J Exp Med 1998;187:517-23.
[4]Ballou LR. The regulation of cyclooxygenase-1 and -2 in knockout cells and mice. Adv Exp Med Biol 2002;507:585-91.
[5]Zhang J,Goorha S,Raghow R,Ballou LR. The tissue-specific,compensatory expression of cyclooxygenase-1 and -2 in transgenic mice. Prostaglandins Other Lipid Mediat 2002;67:121-35.
[6]Bottone Jr FG,Martinez JM,Alston-Mills B,Eling TE. Gene modulation by Cox-1 and Cox-2 specific inhibitors in human colorectal carcinoma cancer cells. Carcinogenesis 2004;25:349-57.
[7]Li H,Hortmann M,Daiber A,Oelze M,Ostad MA,Schwarz PM,et al. Cyclooxygenase 2-selective and nonselective nonsteroidal anti-inflammatory drugs induce oxidative stress by up-regulating vascular NADPHoxidases. J Pharmacol ExpTher 2008;326:745-53.
[8]Rodemann P,Rennekampff HO. Functional diversity of fibroblasts in tumor. In: Mueller MM,Fusenig RZ,editors.Tumor-associatedfibroblasts andtheir matrix. Netherlands: Springer;2011. p. 23-36.
[9]Amin AR,Attur M,Patel RN,Thakker GD,Marshall PJ,Rediske J,et al. Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J Clin Invest 1997;99:1231-7.
[10]Attur M,Dave M,Abramson SB,Amin AR. Activation of diverse eicosanoid pathways in osteoarthritic cartilage: a lipidomic and genomic analysis. Bull NYU Hosp Jt Dis 2012;70:99-108.
[11]Amin AR,Wang G. Identification and characterization of transcriptome-based biomarkers in arthritis and cancer for personalized medicine by translational genomics biomedical mathematics. In: Censor Y,Jiang M,Wang G,editors. Promising directions in imaging,therapy planning and inverse problems. Madison: Medical Physics Publishing;2009. p. 12-20.
[12]MacBeath G,Saghatelian A. The promise and challenge of’-omic’approaches. Curr Opin Chem Biol 2009;13:501-2.
[13]Bonetta L. Protein-protein interactions: interactome under construction. Nature 2010;468:851-4.
[14]Langenbach R,Loftin CD,Lee C,Hiano H. Cyclooxygenasedeficient mice. A summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci 1999;889:52-61.
[15]Langenbach R,Loftin C,Lee C,Hiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem Pharmacol 1999;58:1237-46.
[16]Singh BK,Haque SE,Pillai KK. Assessment of nonsteroidal antiinflammatory drug-induced cardiotoxicity. Expert Opin Drug Metab Toxicol 2014;10:143-56.
[17]Lenzer J. FDA advisers warn: COX 2 inhibitors increase risk of heart attack and stroke. BMJ 2005;330:440.
[18]Lou J,Fatima N,Xiao Z,Stauffer S,Smythers G,Greenwald P,et al. Proteomic profiling identifies cyclooxygenase-2-17 global proteomic changes by celecoxib in colorectal cancer cells. Cancer Epidemiol Biomarkers Prev 2006;15:1598-606.
[19]Trifan OC,Smith RM,Thompson BD,Hla T. Overexpression of cyclooxygenase-2 induces cell cycle arrest. Evidence of a prostaglandin-independentmechanism.JBiolChem 1999;274:34141-7.
[20]Weissmann G,Montesinos MC,Pillenger M,Cronstein BN. Nonprostaglandin effects of aspirin III and salicylate: inhibition of integrin-dependent human neutrophil aggregation and inflammation in COX 2- and NF kappa B(P105)-knockout mice. Adv Exp Med Biol 2002;507:571-7.
[21]Attur MG,Dave MN,Amin AR. Functional genomics approaches in arthritis. Am J Pharmacogenomics 2004;4:29-43.
[22]Dinarello CA. Biologic basis for interleukin-1 in disease. Blood 1996;87:2095-147.
[23]Church LD,Cook GP,McDermott MF. Primer: inflammasomes and interleukin 1β in inflammatory disorders. Nat Clin Pract Rheumatol 2008;4:34-42.
[24]Lukens JR,Gross JM,Kanneganti TD. IL-1 family cytokines trigger sterile inflammatory disease. Front Immunol 2012;3:315.
[25]Chen GY,Nun˜ez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010;10:826-37.
[26]Irizarry RA,Bolstad BM,Collin F,Cope LM,Hobbs B,Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Res 2003;31:e15.
[27]Rao R,Elliott MR,Leitinger N,Jensen RV,Goldberg JB,Amin AR. RahU: an inducible and functionally pleiotropic protein in Pseudomonas aeruginosa modulates innate immunity and inflammation in host cells. Cell Immunol 2011;270:103-13.
[28]Amin AR,Islam BM. Genomics analysis and differential expression of HMG and S100A family in human arthritis: upregulated expression of chemokines,IL-8 and nitric oxide by HMGB1. DNA Cell Biol 2014;33:550-65.
[29]Burke JE,Dennis EA. Phospholipase A2 structure/function,mechanism,and signaling. J Lipid Res 2009;50:S237-42.
[30]Lillig CH,Berndt C. Cellular functions of glutathione. Biochim Biophys Acta 2013;1830:3137-8.
[31]Hayes JD,Flanagan JU,Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 2005;45:51-88.
[32]Zelko IN,Mariani TJ,Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD(SOD1),Mn-SOD (SOD2),and EC-SOD(SOD3)gene structures,evolution,and expression. Free Radic Biol Med 2002;33:337-49.
[33]Bevers EM,Williamson PL. Phospholipid scramblase: an update. FEBS Lett 2010;583:2724-30.
[34]Szklarczyk D,Franceschini A,Kuhn M,Simonovic M,Roth A,Minguez P,et al. The STRING database in 2011: functional interaction networks of proteins,globally integrated and scored. Nucleic Acids Res 2011;39:D561-8.
[35]Ross D,Jadwiga K,Kepa A,Shannon L,Winski A,Howard D,et al. NAD(P)H:quinone oxidoreductase 1(NQO1),chemoprotection,bioactivation,gene regulation and genetic polymorphisms. Chem Biol Interact 2000;129:77-9.
[36]Daniel Nebert W,Karp CL. Endogenous functions of the aryl hydrocarbon receptor(AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem 2008;283:36061-5.
[37]Buczynski MW,Dumlao DS,Dennis EA. An integrated omics analysis of eicosanoid biology. J Lipid Res 2009;50:1015-38.
[38]Lih-Ling L,Lin AY,DeWitt DL. Interleukin-1α induces the accumulation of cytosolic phospholipase A2and the release of prostaglandinE2inhuman fibroblasts. J Biol Chem 1992;267:23451-4.
[39]Font-Nieves M,Sans-Fons MG,Gorina R,Bonfill-Teixidor E,Salas-Pe´rdomo A,Ma´rquez-Kisinousky L,et al. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide(LPS)control prostaglandin E2 production in astrocytes. J Biol Chem 2012;287:6454-68.
[40]Marcouiller P,Pelletier JP,Gue´vremont M,Martel-Pelletier J,Ranger P,Laufer S,et al. Leukotriene and prostaglandin synthesis pathways in osteoarthritic synovial membranes: regulating factors for interleukin 1beta synthesis. J Rheumatol 2005;32:704-12.
[41]Ashok Amin R,Thompson SD,Amin SA. Future of genomics in diagnosis of human arthritis: the hype,hope and metamorphosis for tomorrow. Future Rheumatol 2008;2:385-9.
[42]Eun Choi M,Kim SR,Lee EJ,Han JA. Cyclooxygenase-2 functionally inactivates p53 through a physical interaction with p53. Biochim Biophys Acta 2009;1793:1354-65.
[43]Grosch S,Tegeder I,Niederberger E,Brautigam L,Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J 2001;15:2742-4.
[44]Hayden MS,Ghosh S. NF-κB,the first quarter-century: remarkable progress andoutstandingquestions. Genes Dev 2012;26:203-34.
[45]Gilroy DW,Colville-Nash PR,Willis D,Chivers J,Paul-Clark MJ,Willoughby DA. Inducible cyclooxygenase may have antiinflammatory properties. Nat Med 1999;5:698.
[46]Gomez PF,Pillinger MH,Attur M,Marjanovic N,Dave M,Park J,et al. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-κB subunits(p65,p50)in stimulatedrheumatoidsynovial fibroblasts. J Immunol 2005;175:6924-30.
[47]Degner SC,Kemp MQ,Hockings JK,Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer 2007;59:248-57.
[48]Jared A,Sheridan B,Michela Z,Nair P,Qutayba H,Eidelman DH,et al. The Aryl hydrocarbon receptor attenuates cytoplasmic translocationof HuRandsubsequent COX-2productioninhuman lung fibroblasts. American Thoracic Society 2013 International Conference;Philadelphia Pennsylvania(May 17-22):A2706.
[49]Alexander DL,Ganem LG,Fernandez-Salguero P,Gonzalez F,Jefcoate CR. Aryl-hydrocarbon receptor is an inhibitory regulator of lipid synthesis and of commitment to adipogenesis. J Cell Sci 1998;111:3311-22.
[50]Di Meglio P,Duarte JH,Ahlfors H,Owens ND,Li Y,Villanova F,et al. Activation of the aryl hydrocarbon receptor dampens the severityofinflammatoryskinconditions.Immunity 2014;40:989-1001.
[51]O’Donnell EF,Saili KS,Koch DC,Kopparapu PR,Farrer D,Bisson WH,et al. The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS One 2010;15: e13128.
[52]Thatcher TH,Maggirwar SB,Baglole CJ,Lakatos HF,Gasiewicz TA,Richard RP,et al. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component RelB. Am J Pathol 2007;170:855-64.
[53]Daikoku T,Tranguch S,Trofimova IN,Dinulescu DM,Jacks T,Nikitin AY,et al. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res 2006;66:2527-31.
[54]Sano H,Kawahito Y,Wilder RL,Hashiramoto A,Mukai S,Asai K,et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res 1995;55:3785-9.
[55]Sobrino A,Mata A,Laguna-Fernandez A,Novella S,Oviedo PJ,Garcia-Perez MA,et al. Estradiol stimulates vasodilatory and metabolic pathways in cultured human endothelial cells. PLoS One 2009;4:e8242.
[56]von Moltke J,Trinidad NJ,Moayeri M,Kintzer AF,Wang SB,van Rooijen N,et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012;490:107-11.
[57]Dave M,Amin AR. Yin-Yang regulation of prostaglandins and nitric oxide by PGD2in human arthritis: reversal by celecoxib. Immunol Lett 2013;152:47-54.
[58]Ma K,Langenbach R,Rapoport SI,Basselin M. Altered brain lipid composition in cyclooxygenase-2 knockout mouse. J Lipid Res 2007;48:848-54.
[59]Rhee SG,Kang SW,Chang TS,Jeong W,Kim K. Peroxiredoxin,a novel family of peroxidases. IUBMB Life 2001;52:35-41.
[60]Kruger A,Gruning NM,Wamelink MM,Kerick K,Kirpy A,Parkhomchuk D,et al. The pentose phosphate pathway is a metabolic redox sensor and regulates transcription during the antioxidant response. Antioxid Redox Signal 2011;15:311-24.
[61]Smyth EM,Grosser T,Wang M,Yu Y,FitzGerald GA. Prostanoids in health and disease. J Lipid Res 2009;50:S423-8.
[62]Clancy Robert,Varenika B,Huang W,Ballou L,Attur M,Amin AR,et al. Nitric oxide synthase/COX cross-talk: nitric oxide activates COX-1 but inhibits COX-2-derived prostaglandin production. J Immunol 2000;165:1582-7.
[63]Patel R,Attur MG,Dave D,Abramson SB,Amin AR. Regulation of cytosolic COX-2 and prostaglandin E2 production by nitric oxide in activated murine macrophages. J Immunol 1999;162:4191-7.
[64]de Jonge HJ,Fehrmann RS,de Bont ES,Hofstra RM,Gerbens F,Kamps WA,et al. Evidence based selection of housekeeping genes. PLoS One 2007;2:e898.
[65]Perez-Llamas C,Lopez-Bigas N. Gitools: analysis and visualisation of genomic data using interactive heat-maps. PLoS One 2011;6:e19541.
[66]Franceschini A,Szklarczyk D,Frankild S,Kuhn M,Simonovic M,Roth A,et al. STRING v9.1: protein-protein interaction networks,with increased coverage and integration. Nucleic Acids Res 2013;41:D808-15.
[67]Shannon P,Markiel A,Ozier O,Baliga NS,Wang JT,Ramage D,et al. Cytoscape: a software environment for integrated models ofbiomolecularinteractionnetworks.GenomeRes 2003;13:2498-504.
APPLICATION NOTE
*Corresponding author.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Long Non-coding RNAs in the Cytoplasm
- LVTree Viewer: An Interactive Display for the All-Species Living Tree Incorporating Automatic Comparison with Prokaryotic Systematics
- Similarity Estimation Between DNA Sequences Based on Local Pattern Histograms of Binary Images
- Up-regulation of Long Non-coding RNA TUG1 in Hibernating Thirteen-lined Ground Squirrels
- Call for Papers Special Issue on“Genome Stability’’
- Call for Papers Special Issue on“Computational Cardiology’’