紫金化毒栓的质量标准研究
2016-04-26张丽艳李艳英马晓晴
夏 浇 张丽艳 李艳英 刘 军 马晓晴
1.贵阳中医学院,贵州 贵阳 550002;2.北京因科瑞斯药物研究院有限公司,北京 102209
紫金化毒栓的质量标准研究
夏浇1张丽艳1李艳英2*刘军2马晓晴2
1.贵阳中医学院,贵州贵阳550002;2.北京因科瑞斯药物研究院有限公司,北京102209
【摘要】目的:建立紫金化毒栓的质量标准。方法:采用薄层色谱法(TLC)对栓剂中金银花、连翘、关黄柏、五倍子进行定性鉴别([1]);采用高效液相色谱法(HPLC)对制剂中绿原酸和连翘苷进行定量测定。结果:薄层鉴别专属性强、斑点清晰、分离度好;绿原酸和连翘苷进样量分别在0.07976~1.994 μg(r=0.9998)、0.1202~3.005 μg(r=0.9999)范围内线性关系良好,平均加样回收率(n=6)分别为98.11%、99.17%,RSD分别为0.92%、0.85%。结论:所建立的定性、定量分析方法简便快速、重复性好、准确度高,能够有效控制紫金化毒栓的质量。
【关键词】紫金化毒栓;HPLC;TLC
紫金化毒栓是名老中医临床经验方,根据中医辨证施治理论将金银花、连翘、黄柏等中药合理配伍,并按一定比例配方制成的栓剂[2],具有清热解毒、活血燥湿、消肿生肌的功效。主要用于湿热瘀阻所致的带下病,症见带下量多、色黄或异味、或阴部瘙痒以及宫颈炎高危型人乳头瘤病毒感染(HPV)见上述证候者。为了保证临床用药的安全性、有效性,建立紫金化毒栓中有效成分的定性定量控制方法有至关重要的意义。本实验采用TLC法对处方中的金银花、连翘、关黄柏、五倍子进行定性鉴别,采用HPLC法测定栓剂中的金银花中绿原酸的含量和连翘中连翘苷的含量,为紫金化毒栓临床用药的安全有效性提供了保障,也为紫金化毒栓的质量标准建立奠定基础。
1仪器与材料
1.1仪器LC-2010 AHT高效液相色谱仪(日本岛津);色谱柱 Dikma Diamonsil C18(250mm×4.6mm,5μm),色谱柱Platisil ODS C18(250mm×4.6mm,5 μm);电子分析天平;UV-2450紫外分光光度计(岛津);KQ-250DB型数控超声波清洗器;高速冷冻离心机;紫外分析仪。
1.2材料绿原酸对照品(批号:110753-200413,纯度:96.2%)、连翘苷对照品(批号:110821-201112,纯度:96.8%)、没食子酸对照品(批号:11083-200803,纯度:90.1%)、盐酸小檗碱(批号:110713-201212,纯度:86.7%)、金银花对照药材(批号:121060-200805)、关黄柏对照药材(批号:120937-200506)、五倍子对照药材(批号:121078--200402)、紫草对照药材(新疆紫草)(批号:121430-201103)、连翘对照药材(批号:120908-201218)均购自中国食品药品检定研究院;紫金化毒栓(批号:20130401,20130402,20130403,自制);甲醇、乙腈为色谱纯(迪马公司),水为娃哈哈纯净水,其他试剂均为分析纯;硅胶G板、硅胶H板、硅胶GF254板均为青岛海洋化工厂产品。
2方法与结果
2.1薄层鉴别
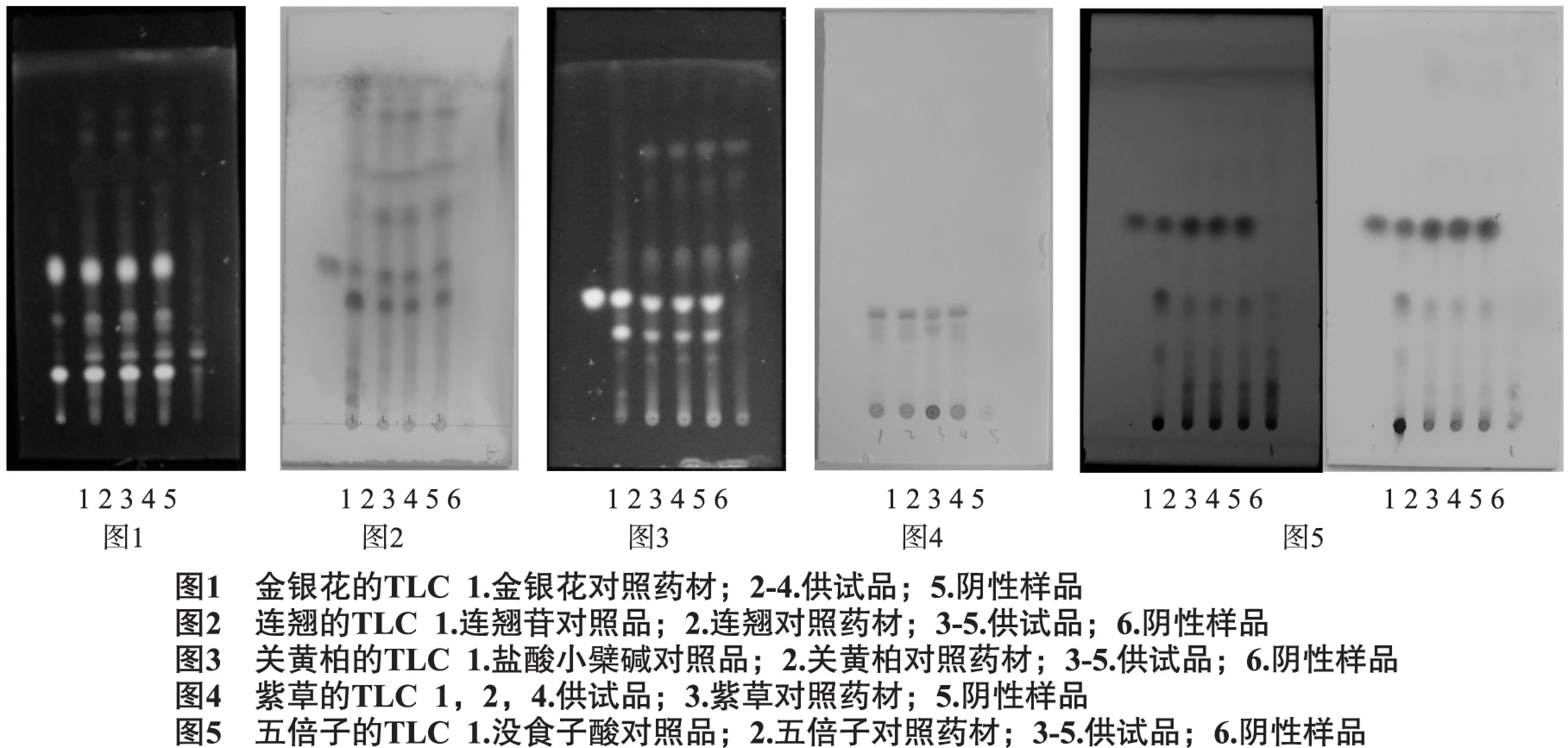
2.1.1金银花的薄层鉴别取本品1粒,置于100ml具塞锥形瓶中,加水30ml,水浴加热使其完全溶散,超声处理30min(40℃),冷冻30min(-20~-10℃),离心,过滤,滤液加入稀盐酸1ml,摇匀,取30ml乙酸乙酯振摇提取,取乙酸乙酯液水浴蒸干,残渣加入甲醇2ml使其溶解,作为供试品溶液。另取金银花对照药材0.5g,加甲醇30ml,超声30min,过滤,滤液浓缩至约2ml,作为对照药材溶液。吸取上述两种溶液各3μl,分别点于同一高效硅胶G薄层板上,以乙酸丁酯-甲酸-水(7∶2.5∶2.5)的上层溶液为展开剂,展开,取出,晾干。置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰,见图1。
2.1.2连翘的薄层鉴别[3]取本品2粒,置于100ml具塞锥形瓶中,加水40ml,水浴加热使其完全溶散,超声处理30min(40℃),冷冻30min(-20~-10℃),离心,过滤,滤液用乙酸乙酯振摇提取3次,每次30ml,合并乙酸乙酯液,水浴蒸干,残渣用70%甲醇5ml使溶解,加于中性氧化铝柱(100~200目,2g,内径1cm,干法装柱),用70%甲醇30ml洗脱,收集洗脱液,蒸干,残渣用甲醇2ml使溶解,作为供试品溶液。另取连翘苷对照品,加甲醇制成每1ml含0.25mg的溶液,作为对照品溶液。再取连翘对照药材1g,加石油醚(30~60℃)20ml,超声处理15min,滤过,弃去石油醚液,残渣挥干,加甲醇20ml,超声处理20min,滤过,滤液蒸干,残渣加甲醇2ml使溶解,作为对照药材溶液。吸取上述三种溶液各5 μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇(20∶3)为展开剂,展开,取出,晾干。喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品和对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰。见图2。
2.1.3关黄柏的薄层鉴别[4]取本品适量,切碎,取约1g,置于100ml具塞锥形瓶中,加50%乙醇25ml,水浴使溶散均匀,40℃超声处理30min,冷冻(-20~-10℃)30min,离心,上清液用少量棉花滤过,滤液蒸干,加甲醇2ml使溶解,滤过,滤液作为供试品溶液。另取盐酸小檗碱对照品适量,加甲醇制成0.2mg·ml-1的对照品溶液。再取关黄柏对照药材0.2g,加甲醇5ml,超声处理30min,滤过,滤液作为对照药材溶液。吸取上述三种溶液各2 μl,分别点于同一硅胶H薄层板,以乙酸丁酯-甲酸-水(7:2.5:2.5)的上层溶液为展开剂,展开,取出,晾干。置紫外光灯(365nm)下检视。供试品色谱中,在与对照品和对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰。见图3。
2.1.4紫草的薄层鉴别[5]取本品1粒,置于100ml具塞锥形瓶中,加无水乙醇25ml,40℃超声处理20min,冷冻(-20~-10℃)30min,离心,取上清液蒸干,残渣加无水乙醇4ml使溶解,用0.45μm的微孔滤膜过滤,滤液作为供试品溶液。另取紫草对照药材0.1g,加70%乙醇回流1h,滤过,滤液蒸干,加无水乙醇25ml,超声处理20min,滤过,滤液浓缩至约4ml,作为对照药材溶液。吸取上述两种溶液各3μl,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,于10℃以下预饱和30min,展开,取出,晾干,日光下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰。见图4。
2.1.5五倍子的薄层鉴别取本品1粒,置于100ml具塞锥形瓶中,加水25ml,水浴使溶散均匀,40℃超声处理30min,冷冻(-20~-10℃)30min,离心,上清液用少量棉花滤过,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取没食子酸对照品,加甲醇制成每1mg·ml-1的溶液,作为对照品溶液。再取五倍子对照药材0.5g,加水25ml,超声处理30min,滤过,滤液蒸干,残渣加甲醇2ml使溶解,作为对照药材溶液。吸取上述三种溶液各2μl,分别点于同一硅胶GF254薄层板上,以三氯甲烷-甲酸乙酯-甲酸(5∶5∶1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。喷以三氯化铁试液,日光下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点,阴性样品无干扰。见图5。
2.2含量测定
2.2.1绿原酸的含量测定
2.2.1.1色谱条件与系统适用性试验
2.2.1.1.1色谱条件以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.4%磷酸(9∶91)为流动相,体积流量 1.0ml/min;检测波长327nm;柱温25℃ ;进样量10 μl。该条件下测出理论板数按绿原酸峰计算应不低于2000,相邻峰分离度均大于1.5且阴性样品无干扰。
2.2.1.1.2对照品溶液的制备取绿原酸对照品适量,精密称定,置棕色容量瓶中,加50%甲醇制成40μg·ml-1的溶液,即得(10℃以下保存)。
2.2.1.1.3供试品溶液的制备取本品,切碎,精密称定0.5g,置具塞锥形瓶中,加硅藻土1.0g,精密加水50ml,称定重量,置60℃水浴5min使溶散均匀,取出,40℃超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用水补足减失的重量,摇匀,冷冻(-20~-10℃)30min,取出,离心10min(转速为4000rpm),取上清液,滤过,即得。
2.2.1.1.4测定方法分别精密吸取对照品溶液与供试品溶液各10 μl,注入液相色谱仪,测定,即得。
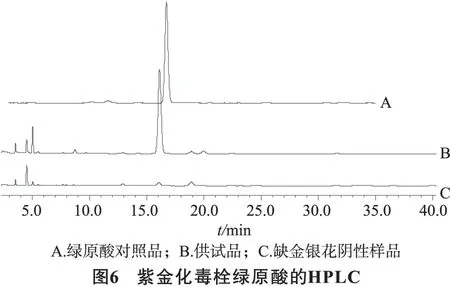
2.2.1.1.5专属性试验除金银花外,按处方比例称取药材及辅料按2.2.1.1.3项下制成阴性样品,再按供试品制备方法,制备成阴性对照溶液,进行测定。结果表明:阴性样品色谱中在与绿原酸相应的保留时间上有色谱峰 ,但计算峰面积约为供试品峰面积的3.56%,小于5%,表明其他成分对绿原酸的测定干扰比较小,绿原酸峰的理论板数n大于2000。样品中绿原酸色谱峰与相近峰分离完全,分离度均大于1.5。本品中绿原酸的含量具有专属性。见图6。
2.2.1.2方法学考察
2.2.1.2.1线性关系的考察精密称取绿原酸对照品9.97mg,置于25ml棕色容量瓶中,加入50%甲醇适量使其完全溶解并定容至刻度,摇匀,制得绿原酸对照品浓度为0.3988mg/ml;再精密量取0.2、0.5、1.0、2.0、3.0、5.0ml分置10ml棕色量瓶中,用50%甲醇稀释至刻度,摇匀,配制成7.976、19.94、39.88、79.76、119.64、199.4μg/ml的对照品溶液,摇匀,分别精密吸取10 μl,注入液相色谱仪进行测定。回归方程:Y=3139590.3720X-95257.0154(r=0.9998)。结果表明绿原酸含量在79.76~1994μg范围内线性关系良好。
2.2.1.2.2精密度试验取本品切碎,精密称定0.5g,按2.2.1.1.3项下方法制备供试品,分别精密吸取10μl,注入液相色谱仪,进行测定,同一供试品连续进样6次。结果绿原酸的峰面积RSD为0.13%,表明仪器精密度良好。
2.2.1.2.3稳定性试验取本品切碎,精密称定0.5g,按2.2.1.1.3项下方法制备供试品,分别于0、2、4、8、12、24h精密吸取10 μl,注入液相色谱仪进行测定,结果RSD为0.21%,表明供试品溶液在24h内稳定性良好。
2.2.1.2.4重复性试验取本品切碎,精密称定6份,每份0.5 g,按2.2.1.1.3项下方法制备供试品,分别精密吸取10μl,注入液相色谱仪进行测定。结果RSD值为0.12%,RSD<2.0%,表明本方法的重复性良好。
2.2.1.2.5加样回收率试验采用加样回收法,精密称取绿原酸对照品8.70mg,置10ml量瓶中,加50%甲醇使溶解并稀释至刻度,摇匀,配制成0.87mg/ml的对照品溶液,精密吸取1ml(共6份),分置不同锥形瓶中,蒸干,然后取本品切碎,精密称定6份,每份0.25g,分置上述锥形瓶中,分别加硅藻土1.0 g,精密加水50 ml,密塞,称定重量,置60℃水浴5 min使溶散均匀,取出,40℃超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用水补足减失的重量,摇匀,冷冻(-20~-10℃)30min,取出,离心10min(转速为4000r/min),微孔滤膜滤过,取续滤液,即得。结果绿原酸含量:3.3936mg/g,平均回收率为98.11%,RSD为0.92%。表明本方法具有较好的回收率。
2.2.1.2.6样品的含量测定取三批样品,按供试品溶液方法制备,按上述色谱条件测定,计算,得到三批样品绿原酸的含量为:7.58、7.58、7.60mg/粒。根据三批样品测定结果及所用的金银花药材中的绿原酸的含量,折算本品连翘苷的提取率均为55%左右;因此本品按2015年版《中国药典》一部“金银花”药材项下绿原酸的含量限度1.5%折算制剂的理论含量为12.85mg/粒,结合实际大生产情况,按50%转移率折算,暂定本品每片含金银花以绿原酸(C16H18O9)计,不得少于6.5mg/粒。
2.2.2连翘苷的含量测定
2.2.2.1色谱条件与系统适用性试验
2.2.2.1.1色谱条件以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(24∶76)为流动相,流量 1.0ml/min,检测波长为230nm,柱温40℃。理论板数按连翘苷峰计算应不低于3000,相邻峰分离度均大于1.5且阴性样品无干扰。
2.2.2.1.2对照品溶液的制备取连翘苷对照品适量,精密称定,加甲醇制成1μg/ml的溶液,即得。
2.2.2.1.3供试品溶液的制备取本品,切碎,取约2g,精密称定,置具塞锥形瓶中,加硅藻土2g,精密加入50%乙醇50ml,称定重量,置60℃水浴15min使溶散均匀,取出,40℃超声处理30min,放冷,再称定重量,用50%乙醇溶液补足减失的重量,摇匀,置于冰箱冷冻层(-20~-10℃)30min,取出,离心(转速4000r/min)10min,滤过,精密量取续滤液25ml,置分液漏斗中,用三氯甲烷振摇提取5次,每次30ml,合并三氯甲烷提取液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.2.2.1.4测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,进行测定。
2.2.2.1.5专属性试验除连翘药材外,按处方比例称取药材及辅料按2.2.2.1.3项下制成阴性样品,进行测定。结果表明阴性样品色谱中在与连翘苷相应的保留时间上无色谱峰,表明其他成分对本品中连翘苷的含量测定无干扰,连翘苷的理论板数n大于3000。样品中连翘苷色谱峰与相近峰分离完全,分离度均大于1.5。以本法测定本品中连翘苷的含量具有专属性。见图7。

2.2.3方法学考察
2.2.3.1线性关系的考察精密称取连翘苷对照品7.76mg,置25ml量瓶中,加甲醇适量超声处理(功率250W,频率40kHz)使溶解,取出,放冷,加甲醇稀释至刻度,摇匀,精密量取0.2、0.5、1.0、2.0、3.0、5.0ml分置10ml量瓶中,用甲醇稀释至刻度,摇匀,配制成6.01、15.02、30.05、60.1、90.15、150.25μg/ml的对照品溶液,摇匀,分别精密吸取20μl,注入液相色谱仪进行测定,得回归方程:Y=2209920.5724X-2526.4149(r=0.9999)。结果表明,连翘苷进样量在0.1202~3.005μg范围内线性关系良好。
2.2.3.2精密度试验取本品切碎,精密称定2g,按2.2.2.1.3项下方法制备供试品,分别精密吸取20μl,注入液相色谱仪进行测定,同一供试品连续进样6次,结果RSD为0.76%。表明该方法的仪器精密度良好。
2.2.3.3稳定性试验取本品切碎,精密称定2g,按2.2.2.1.3项下方法制备供试品,分别于0、2、4、8、12、24h精密吸取10μl,注入液相色谱仪进行测定。RSD为0.72%,表明供试品溶液在24h内稳定性良好。
2.2.3.4重复性试验取本品切碎,取约2g(共6份),按2.2.2.1.3项下方法制备供试品,分别于0、2、4、8、12、24h精密吸取10μl,注入液相色谱仪进行测定。结果RSD为0.72%,小于2.0%,表明该方法的重复性良好。
2.2.3.5加样回收率试验采用加样回收法,精密称取连翘苷对照品14.95mg,置25ml量瓶中,加甲醇使溶解并稀释至刻度,摇匀,配制成0.5789mg/ml的对照品溶液;精密吸取4ml置10ml量瓶中,用甲醇定容至刻度,摇匀,配制成231.56μg/ml的对照品溶液,分别精密吸取1.5ml(共6份),分置不同锥形瓶中,蒸干,再取本品切碎,取约1g(共6份),精密称定,分置上述锥形瓶中,分别加硅藻土2g,精密加入50%乙醇50ml,密塞,称定重量,置60℃水浴15min使溶散均匀,取出,40℃超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用50%乙醇溶液补足减失的重量,摇匀,置于冰箱冷冻层(-20~-10℃)30min,取出,离心(转速4000r/min)10min,滤过,精密量取续滤液25ml,置分液漏斗中,用三氯甲烷振摇提取5次,每次30ml,合并三氯甲烷提取液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,用微孔滤膜滤过,取续滤液,即得。结果连翘苷含量:0.3223mg/g,平均回收率为99.17%,RSD为0.85%。说明本方法具有较好的回收率。
2.2.3.6样品的含量测定取三批样品,按供试品溶液方法制备,按上述色谱条件测定,计算,得到三批样品连翘苷的含量为:0.91、0.93、0.92mg/粒。根据三批样品中连翘苷的测定结果及所用的连翘药材中的连翘苷的含量,折算本品连翘苷的提取率均为50%左右;因此本品按2015年版《中国药典》一部“连翘”药材项下连翘苷的含量限度0.15%折算制剂的理论含量为1.28mg/粒,按50%转移率折算,暂定本品每片含连翘以连翘苷(C27H34O11)计,不得少于0.65mg/粒。
3讨论
3.1紫金化毒栓为未在国内上市销售的中药六类复方制剂,为保证成品质量,对方中药材的主要特征成分进行了鉴别试验。本实验所建立的质量标准薄层鉴别专属性强,斑点清晰、快速、简便、重现性好、分离度好;利用HPLC法对样品中绿原酸、连翘苷进行测定具有操作简单、重复性好、分析时间短、分离效果好等优点,能很好的用于紫金化毒栓的质量控制,保证临床用药的安全、有效。
3.2该栓剂基质为脂溶性基质,薄层色谱鉴别中供试品都进行低温冷冻处理使油脂分离,排除基质对实验结果的干扰。连翘药材提取为水提,故连翘的鉴别中用水作为提取溶剂,但结果背景较深,且分离度不好,需进一步纯化样品,参考《中国药典》连翘苷含量测定方法,增加过中性氧化铝柱的步骤结果排除了背景杂质的影响,显色斑点清晰可见,故确定为本实验的薄层鉴别方法。金银花参照药典中金银花药材的薄层鉴别条件结果,供试品溶液中含栓剂基质太多,无法点样,因此加入盐酸进一步纯化,最后用乙酸乙酯萃取,除去了基质中干扰杂质。
3.3本复方中由于连翘、金银花为君药,其含有的成分连翘苷、绿原酸均具有抗菌作用,因此确定连翘苷、绿原酸作为本品的含量测定成分。金银花加样回收试验样品制备过程加入硅藻土,吸附了基质中的油脂,排除其对含量的影响,以便更好地控制产品质量。
参考文献
[1]国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2015.
[2]袁婕,李婧,黄松,等.妇科栓剂的质量标准研究[J].中国实验方剂学杂志,2010,16(14):68-70.
[3]夏伯候,朱晶晶,王志民,等.连翘药材新增定量标准研究[J].中国中药杂志,2010,35(16):72-74.
[4]陈丽卿.黄柏与关黄柏的薄层鉴别[J].医学前沿,2014,7(5):55-56.
[5]王彩琴,苏亚宁,王巧明,等. 高效液相色谱法测定复方紫冰栓中左旋紫草素的含量[J].中国医院药学杂志,2009,29(12):1053-1054.
实验研究
Study on quality standard for Zijin Huadu Suppository
XIA Jiao1ZHANG Liyan1LI Yanying2*LIU Jun2MA Xiaoqing2
1.Guiyang College of Traditional Chinese Medicine,guiyang 550002,China;2.Beijing Increase Pharmaceutical Institute Co.,Ltd. beijin 102209,China
Abstract:Objective To establish the quality standard of the Zijin Huadu suppository. Methods Exert qualitative identification of Honeysuckle,Forsythia,Cortex Phellodendri, Galla chinensis by Thin Layer Chromatography(TLC) and quantitative identification of chlorogenic acid and Phillyrin by High Performance Liquid Chromatography(HPLC).Results TLC identification shows great specificity,that spots are clear and resolution is good. When the injection volume of chlorogenic acid and forsythin are within the range respectively: 0.07976-1.994 μg(r=0.9998),0.1202-3.005 μg(r=0.9999),the linearity is favorable. The average recovery rate(n=6) of chlorogenic acid and forsythin are 98.11% and 99.17% while the RSD are 0.92% and 0.85%.Conclusion The qualitative and quantitative identification methods we established are simple and fast and have good repeatability and accuracy, by which we can effectively control the quality of Zijin Huadu Suppository.
Key words:Zijin Huadu Suppository;HPLC; TLC
(收稿日期:2015.12.31)
【中图分类号】R286
【文献标志码】A
【文章编号】1007-8517(2016)06-0030-04
作者简介:夏浇,硕士,主要从事中药学质量控制研究。E-mail:1069511329@qq.com。通信作者:李艳英,硕士,工程师,主要从事新药研究。E-mail:yanyingliabc@163.com 。
基金项目:北京市中医局科技发展资金项目(JJ2014-15)。
