常压下氢氧直接合成过氧化氢用钯催化剂研究进展
2016-04-12田敏潘红艳林倩陈志超赵敏陈政
田敏,潘红艳*,林倩,陈志超,赵敏,陈政
(1.贵州大学化学与化工学院,贵州贵阳550025;2.贵州省绿色化工与清洁能源技术重点实验室,贵州贵阳550025)
常压下氢氧直接合成过氧化氢用钯催化剂研究进展
田敏1,潘红艳1*,林倩2*,陈志超1,赵敏1,陈政1
(1.贵州大学化学与化工学院,贵州贵阳550025;
2.贵州省绿色化工与清洁能源技术重点实验室,贵州贵阳550025)
综述了常压下氢氧直接合成过氧化氢用钯催化剂研究进展,包括单一负载型钯催化剂、钯催化剂活性组分改性(金属及非金属的掺杂改性)和载体改性(酸改性及其他方法改性)的研究状况。讨论了催化剂活性组分钯颗粒尺寸、分散度、能量位点以及载体表面酸性对氢氧直接合成过氧化氢活性的影响。指出强化主反应,抑制副反应,以提高过氧化氢收率和浓度,达到工业化应用的需求,仍是钯催化剂研究的发展方向。
过氧化氢;直接合成;钯催化剂;改性
氢氧直接合成过氧化氢(H2O2)自上世纪以来一直是绿色化学领域追逐的热点,其研究最早可追溯到1914年[1],早期报道中,由于合成的H2O2浓度极低,研究处于停滞状态,直到20世纪80年代后期,Du Pont公司一份专利中报道,在酸性溶液中,反应温度7~15℃、压力11.4MPa时,以Pt/Pd为催化剂合成H2O2的质量分数达17%[2],这才引起研究者的广泛关注。但过高的反应压力,增加了H2、O2爆炸的危险性,限制了该工艺的应用。近三十年来,为寻求温和反应条件下H2O2的高浓度和高收率,科学家一直围绕Pt、Pd及相关的催化剂进行研究。
在近年来直接合成H2O2的相关研究中,研究者常将以Pd为活性组分、多孔材料为载体制备的钯(Pd)催化剂作为直接合成H2O2的主要催化剂[3-5]。其原因在于贵金属Pd外层电子排布为4d105s0,在一定外界条件下,d轨道电子可跃迁到S轨道,形成d带空穴,产生化学吸附,利于活化H2和O2。据报道,H2经Pd活化后,H-H键解离所需活化能较低(为4.587kJ/mol[6],也有报道指出不需要活化能[7]),H-H键较易断裂,在Pd活性位上主要以吸附态氢原子(Ha)形式存在;O2经Pd活化后,O-O键解离所需活化能较高(有报道认为是49.404kJ/mol[8],也有报道认为是84.992kJ/mol[9],甚至更大),O-O键较难断裂,在Pd活性位上主要以吸附态分子氧(O2)a形式存在。故H2和O2的反应历程通常被认为是Ha、(O2)a在Pd上先合成过渡态过氧物种(OOH)a,再经反应合成H2O2[10-11]。
但当Pd活性位提供的能量达到O2或(OOH)a中O-O键的解离能时,O-O键断裂形成吸附态原子氧2Oa,2Ha和Oa经催化合成H2O,降低H2O2选择性[12];同时,H2O2性质不稳定,自身易发生分解反应(H2O2→H2O+H2),当与H2碰撞时,可能会发生氢化反应(H2O2+H2→H2O)。涉及的反应机理可用图1表示[12-13]。
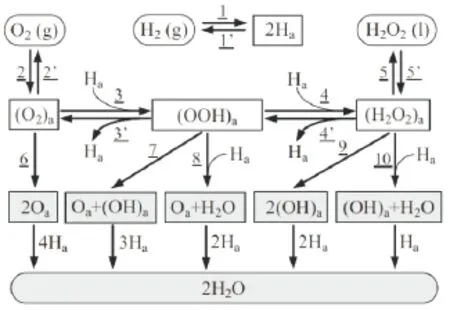
图1 H2和O2直接合成H2O2反应机理及可能的副反应路径(“a”代表“吸附”)
基于氢氧直接合成H2O2反应机理,研究者常从调节活性组分与多孔载体性质两个方面探讨对催化剂性能的影响。通过对活性组分Pd和多孔载体改性,在抑制O2/(OOH)a的O-O键解离,抑制H2O2分解/氢化、选择性合成H2O的副反应,提高H2O2稳定性和选择性的同时,提高活性组分分散度,以提高反应物H2和O2在活性位上的吸附与反应速率,提高催化剂活性。研究方法主要集中在金属、非金属改性活性组分,酸及其他方法改性多孔载体等方面。本文从Pd催化剂活性组分和载体的物化性质对其合成H2O2活性的影响入手,对Pd催化剂活性组分和载体的改性方法进行综述,为后续过氧化氢直接合成催化剂的研制提供借鉴。
1 钯催化剂活性组分和载体对氢氧直接合成H2O2活性的影响
1.1 钯催化剂活性组分的影响
活性组分Pd对H2O2直接合成性能的影响主要有Pd颗粒尺寸、分散度、晶体晶面能量等因素,各因素间彼此独立又相互影响。分散度(D)与金属粒子大小(d/nm)呈反比关系(D=1/d)[14]。按催化活性中心学说观点,认为活性金属颗粒尺寸越小,分散度越高,提供的活性位点越多,单位催化剂、单位时间反应物转化率越大,催化活性越好。如Ye等[15]制备Pd颗粒尺寸分别为3nm、6.5nm和9.5nm的Pd/C催化剂,进行H2O2直接合成活性测试,结果指出反应6h后,Pd/C(3nm)合成H2O2收率为16.2%,而Pd/C(9.5nm)合成H2O2收率则低至1.8%。
也有部分研究者报道,Pd颗粒尺寸低至1nm~2nm时,虽然分散度很高,提供的活性位点较多,但Pd晶面的表面能量较高,容易活化O2中O-O键,使其断裂,形成吸附态原子氧2Oa,与2Ha经催化合成H2O,降低合成H2O2的选择性;此外,也容易活化过渡态过氧物种(OOH)a的O-O键,降低H2O2稳定性。如Menegazzo等[16]制备Pd颗粒尺寸分别为1nm和4nm的2.5Pd/ZrO2和1.5Pd500/SiO2催化剂,反应5h后,合成H2O2的选择性分别约为48%和60%。Ghedini等[17]制备Pd的颗粒尺寸分别为2nm、2nm和4.5nm的1.5Pd/SiO2、1.5Pd/MCM-41和1.5Pd/SBA-15催化剂,反应5h后,Pd合成H2O2的转换频率(TOF)分别是762mmol·g-1·h-1、479mmol·g-1·h-1、1134mmol·g-1·h-1。
Tian和kim等进一步分析Pd的晶面(Pd(111)、Pd(100)和Pd(110))对H2O2直接合成性能的影响。如Tian等[18]利用密度泛函理论(DFT)计算H2和O2在Pd三个晶面上的吸附反应,发现Pd(110)和Pd(100)的晶面能量较Pd(111)高,容易活化O2中O-O键,使其断裂,形成吸附态原子氧2Oa,与2Ha合成H2O,降低H2O2选择性,得到Pd(111)晶面对H2O2直接合成起主要作用的结论;为降低Pd的晶面能量,抑制O-O键的解离,该作者提出可向Pd催化剂中添加活性较小的Au、Ag和Cu等。Kim及其团队[19-20]制备不同晶面Pd晶体,利用实验数据论证Pd晶面与催化活性的关系。如采用胶体法制备Pd(100)晶面的纳米立方体单晶Pd[19],指出Pd(100)晶面催化H2O2生成的同时促进副反应H2O2的氢化和分解的发生,降低H2O2选择性。同样采用胶体法制备Pd (100)晶面的立方体单晶Pd和Pd(111)晶面的纳米八面体单晶Pd[20],指出Pd(111)晶面合成H2O2的选择性和收率均高于Pd(100)晶面,作者认为这是因为Pd(100)晶面更易使O2、OOH和HOOH中O-O键解离,这是生成H2O的关键因素。
1.2 载体微观结构和表面化学性质的影响
载体微观结构和表面化学性质对H2O2直接合成性能的影响主要有载体的晶型、孔径分布、表面酸性(酸量、酸种类)等因素。载体微观结构影响活性组分在载体表面分布的颗粒尺寸及分散度,影响H2O2选择性;表面酸性影响H2O2分解速率,影响H2O2稳定性。
载体微观结构的影响。Hua[21]等以VGCF和XC-72为载体制备Pd/VGCF和Pd/XC-72催化剂,研究发现,两组催化剂上活性组分Pd颗粒尺寸分别为4.3nm和5.0nm,但前者催化剂合成H2O2选择性和产率分别为42%和51mmol·g-1·h-1,后者催化剂增加到74%和129mmol·g-1·h-1,作者认为这是由于VGCF碳材料中仅含有(002)石墨结构,XC-72碳材料中含有(002)和(100)石墨结构,XC-72的高度石墨结构提高其合成H2O2选择性和产率。Pashkova等[22]分别以TiO2和C为载体制备Pd催化剂用于H2O2直接合成,研究发现,2%Pd/TiO2和2%Pd/C催化剂上活性组分Pd的颗粒尺寸分别为2.5nm和19.2nm,合成H2O2选择性分别为79%和69%,Pd合成H2O2的TOF分别为0.93mmol·g-1·h-1和0.78mmol·g-1·h-1。Lee等[23]分别以Al2O3和SiO2为载体制备Pd/Al2O3和Pd@SiO2催化剂,研究发现,两组催化剂上活性组分Pd分散度分别为13%和44%,Pd合成H2O2的TOF分别为253mmol·g-1·h-1和554mmol·g-1·h-1。
载体表面酸性的影响。Park等[24]以HZSM-5-X(X是硅铝比,为15、30、75、100和150)为载体制备Pd催化剂用于H2O2直接合成,研究发现,催化剂表面B酸对H2O2直接合成起关键作用,质子B酸的存在抑制H2O2的分解,提高H2O2的稳定性,使得H2O2收率随B酸与L酸比值(B/L)的增加而增大,其中硅铝比为30的Pd/HZSM-5-30催化剂表面B酸量最大(吸附NH3量5.1mol/g),催化活性最好。该研究团队以TiO2-ZrO2(TZ-X,X=n(ZrO2)/n(TiO2)×100= 0,25,50,75,100)为载体[25],利用等体积浸渍法制备Pd催化剂,发现Pd/TZ-75表面酸量及总酸量最大,合成H2O2收率也最高。
综上可知,Pd催化剂活性组分和载体性质都影响其直接合成H2O2的选择性和稳定性。其中,Pd的高度分散是直接合成H2O2的首要条件,但Pd分散度过高,颗粒尺寸过小,Pd位点能量较高,容易活化O2和(OOH)a中O-O键,使其解离,降低合成H2O2的选择性和稳定性。此外,载体性质也会影响活性组分Pd的分散度和颗粒尺寸,影响H2O2的选择性,其表面酸性还会影响H2O2的稳定性。为了协同它们之间关系,研究者针对Pd催化剂活性组分和载体进行了大量改性研究。
2 活性组分改性对反应活性的影响
2.1 贵金属对钯催化剂活性组分改性
为了提高Pd分散度的同时降低Pd位点能量,协同两者之间的关系,以提高H2和O2在活性位上的吸附与活化、抑制O2和(OOH)a中O-O键解离,从而提高H2O2选择性。研究者以Pd为主活性组分,通过添加第二种金属(如Ag[26-27]、Pt[27-29]、Rh[28]、Ru[28]、Au[30-31]等)制备复合贵金属催化剂,用于H2O2直接合成。表1列出了近年来一些复合贵金属Pd催化剂(主要是Au和Pt的掺杂改性)用于H2O2直接合成的研究。
由表1可见,经Au或Pt改性后的复合贵金属Pd催化剂合成H2O2选择性、收率或产率明显高于单一贵金属Pd催化剂。如Edwards[38]以w(Pd)=5%的Pd/Al2O3和w(Au)=2.5%、w(Pd)=2.5%的Au-Pd/Al2O3为催化剂直接合成H2O2,产率分别是12mmol·g-1·h-1和18mmol·g-1·h-1。Edwards[39]以Al2O3、α-Fe2O3、TiO2、SiO2、活性C为载体,Au、Pd、Au-Pd为活性组分制备催化剂,所得催化剂活性大小皆为Au/Pd>Pd> Au。
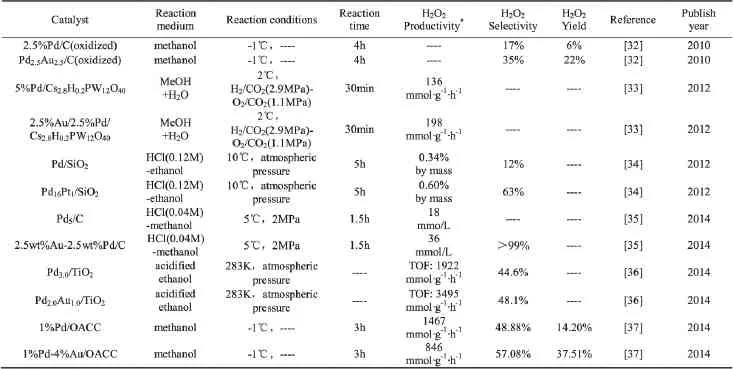
表1 近期文献中研究的贵金属掺杂对Pd催化剂直接合成H2O2性能的影响
为了了解Pt或Au掺杂到Pd催化剂中提高其催化活性的原因,Xu等[34]利用Pt改性Pd催化剂,指出少量Pt掺杂后的Pd16Pt1催化剂,减弱了Pd-O键的结合能,使其容易断裂,同时抑制吸附态O2中O-O键解离,增加了生成OOHad的浓度,提高了H2O2的反应速率(OOHad+H→H2O2),使得Pd16Pt1/SiO2催化剂合成H2O2产率及选择性分别提高到0.60%(质量分数,下同)和63%,而Pd/SiO2催化剂的仅为0.34%和12%。Pritchard等[40]以炭为载体,采用固溶胶法制备不同Au/Pd比的Au-Pd负载型催化剂用于H2O2直接合成,发现Pd/C催化剂上Pd的平均颗粒尺寸为5.7nm,反应30min后,H2O2产能120mmol·g-1·h-1;掺杂Au后,AuPd/C催化剂上Pd的平均颗粒尺寸都下降,其中m(Au):m(Pd)=1:2制备的催化剂上Pd的颗粒尺寸为2.9nm,催化活性最好,相同条件下得到H2O2产能达188mmol·g-1·h-1。Li等[6]对H2O2在Pd和Pd@Au表面的合成机理进行了研究,提出主反应与副反应的竞争主要是O-O键和O-M(M代表Pd(111)面的Pd和Pd@Au表面的Au)键之间的竞争,并指出O-Pd键键能强于中间物OOH和H2O2中O-O键键能,而O-Au键键能反而弱于O-O键键能,使得Au添加到Pd中能弱化OPd键键能,使其易于断裂,相反,也就抑制了O-O解离,即在Pd@Au表面更易合成H2O2且H2O2更容易脱附,从而提高H2O2选择性,增强了H2O2产率。
可见,Au或Pt的改性,降低了活性组分Pd的颗粒尺寸或减弱了Pd-O键键能,抑制吸附态分子氧(O2)a解离为吸附态的原子氧Oa,抑制合成H2O的反应,增加合成H2O2的选择性。
2.2 非贵金属对钯催化剂活性组分改性
部分研究者利用非贵金属,如过渡金属(Ce、Co、Ni、Cr、Mn、Zn、Cd、Cu等)、非金属(卤素离子(Cl-、Br-))对Pd催化剂活性组分进行掺杂改性。如安红强等[41]利用过渡金属M(M=Ce、La、Pt、Fe、Co、Ni、Cr、Mn、Zn、Cd、Cu)对Pd/TS-1掺杂改性,制备系列催化剂用于H2O2直接合成,研究发现,与Pd/TS-1相比,经过渡金属改性后的催化剂都不同程度提高了H2O2收率,其中,经Ce改性后的Pd-Ce/TS-1上Pd的颗粒尺寸由改性前的5nm减小为3nm,分散度增加,提高其合成H2O2的收率。杜旭等[42]采用等体积浸渍法制得过渡金属改性的系列催化剂MOx-PdO/ γ-Al2O3(M=La、Mn、Ni、Ce)用于H2O2直接合成,得到类似的结论,即过渡金属M(M=La、Mn、Ni、Ce)改性可提高PdO/γ-Al2O3催化剂催化H2、O2直接合成H2O2的活性。
非金属改性方面,笪国进等[43]利用吡啶改性Pd/ SiO2催化剂用于H2O2直接合成,指出催化剂经吡啶改性后,提高其合成H2O2产率。此外,Choudhary团队[44-45]发表了较多关于利用卤素离子(F-、Cl-、Br-、I-)对Pd催化剂进行掺杂改性的研究报道,他们认为卤素离子的电负性会明显影响Pd催化剂的催化活性,其中负电性最强的F-不能抑制H2O2的快速分解,负电性最弱的I-则会使催化剂强烈中毒,而具有中间电负性的Cl-或Br-一方面会抑制O2和H2O2中O-O键的断裂,减弱生成H2O的选择性,提高H2O2的稳定性和选择性,另一方面,Br-离子吸附在催化剂表面,改变了活性金属的电子密度,使得金属表面呈现缺电子状态,和O2相比,更利于吸附H2,提高H2O2的选择性。如利用Br-、Cl-、F-改性w(Pd)=2.5%的Pd/Ga2O3,发现Br-改性前后催化剂合成H2O2的收率分别为5.4%和24.8%,H2O2的选择性分别为10.8%和64.5%,而掺杂F-、Cl-时,H2O2的收率和选择性均降低[46]。该团队进一步探讨了双卤素改性Pd催化剂,通过对比F-Br-Pd/Al2O3和Pd/Al2O3在H2O2直接合成的研究发现,反应0.25h后,前者合成H2O2收率是75%,而后者反应1h后,H2O2收率也才达到50%[47]。研究还发现,卤素离子的添加顺序也会影响催化活性,实验中催化活性大小为:F-Br-Pd/ Al2O3(先浸渍Br后浸渍F)>Br-F-Pd/Al2O3(先浸渍F后浸渍Br)>Br/F-Pd/Al2O3(同时浸渍F和Br),其中F(0.13)-Br(0.13)-Pd/Al2O3合成H2O2的选择性和收率都达到78%,H2转化率达到100%[48]。
3 载体改性对催化活性的影响
3.1 酸对钯催化剂载体改性
前人研究发现,反应介质中H2O2稳定性较差,会发生分解反应和氢化反应[49-50]。为此,研究者针对反应介质进行了较多的研究,经研究发现,H2O2在酸性介质中稳定性较强[51-52],但酸性会溶解部分活性组分Pd,同时也会增加后续H2O2分离提纯的难度。鉴于此,研究者利用酸对载体进行改性,将一些酸性官能团,如磺酸基(SO3H)嫁接到多孔载体制备固体超强酸催化剂、或直接以杂多酸为载体制备催化剂,以期提高H2O2稳定性。
研究者发现利用酸对载体进行改性,不仅能提高H2O2稳定性,还能改变活性组分Pd的颗粒尺寸及分散度。如Edwards等[53-54]采用HNO3(w=2%)预处理载体TiO2和SiO2后负载Pd和Pd-Au制备催化剂,发现催化剂活性组分颗粒尺寸减小,合成H2O2的选择性和产率增加。该研究者发表在Science上的文章[55]指出,利用酸改性活性炭(C)制备Au-Pd/C催化剂后,Au-Pd合金纳米颗粒粒径减小,抑制H2O2分解,提高H2O2选择性和收率;其中当Au、Pd质量分数均为2.5%,经乙酸(w=2%)预处理C后制备的催化剂效果最好,H2O2选择性和单位生产能力分别达到98%和175mmol·g-1·h-1,与未经酸预处理的2.5%Au-2.5%Pd/C催化剂相比,分别提高了22.5%和59%。
也有研究者发现通过嫁接酸性基团到多孔载体或者直接以固体超强酸为载体制备催化剂,可明显提高H2O2的稳定性。如Park等[56]以磺酸基(SO3H-)为酸源,利用接枝法将SO3H-接枝到载体SBA-15、MCM-41、MCM-48、MCF、MSU-1上,形成新型的固体超强酸载体,制备系列Pd催化剂,研究发现,与原始催化剂(Pd/SBA-15、Pd/MCM-41、Pd/MCM-48、Pd/MCF、Pd/MSU-1)相比,经SO3H-改性制备的系列催化剂活性都明显提高,催化活性随其表面酸密度的增加而增大,这是因为载体上的酸性基团可作为一种有效酸源抑制H2O2的分解。
为了论证此现象,该团队分别以H3PW12O40、Cs改性的H3PW12O40以及Cs2.5H0.5PW12O40为酸源,对多孔材料MCF进行改性,作为载体分别制备系列Pd催化剂Pd/HPW-MCF-X(X=w(H3PW12O40)×100,分别为1.0,4.8,9.1,13.0,16.7,20.0,23.1,25[57])、Pd/CsxPWMCF(X=w(Cs)×100,分别为1.7,2.0,2.2,2.5,2.7)[58]、Pd/CsPW-MCF-X(X=w(Cs2.5H0.5PW12O40)×100,分别为14.3,21.8,28.1,33.4,38)[59],用于H2O2直接合成,也得到了催化活性随催化剂表面酸密度的增加而增大的结论。
3.2 其他方法改性钯催化剂载体
除利用酸对载体进行改性外,研究者也利用一些非金属如卤素离子(F-、Cl-、Br-)、N元素和碱金属离子对载体进行改性制备Pd催化剂。
Choudhary等[60]利用卤素离子改性γ-Al2O3,制备X/Pd/γ-Al2O3(X=F-、Cl-、Br-)催化剂,研究发现,Br-改性,抑制O2和H2O2中O-O键解离,抑制了H2O2分解和氢化,提高其催化活性,与Pd/γ-Al2O3催化剂相比,Br/Pd/γ-Al2O3催化剂合成H2O2选择性和收率分别从34.8%和8.6%提高至81.8%和17.1%。
Abate等[61]利用N改性多孔碳纳米管制备Pd催化剂,用于H2O2直接合成,研究发现,与Pd/CNT催化剂相比,Pd/N-CNT催化剂上活性组分Pd的颗粒尺寸由原来的3.6nm下降到2.7nm,合成H2O2的选择性和收率分别由原来的35%和98mmol·g-1·h-1提高到45%和128mmol·g-1·h-1。
Park等以不溶性杂多酸为载体制备Pd催化剂(Pd0.15M2.5H0.2PW12O40(M=K,Rb,Cs))[62],利用碱金属离子改性载体,研究发现,碱金属离子(=K,Rb,Cs)掺杂,改变了Pd的电子状态,H2O2的收率随催化剂中Pd 3d5/2结合能的增加而增大,其中Cs改性的催化剂Pd-CsPW中Pd 3d5/2结合能最大,该催化剂上获得的H2O2的收率也最高。可见,Pd催化剂载体的影响是较为复杂的,其影响机理有待于进一步研究。
4 结语
经过学者们近百年、尤其是21世纪以来的研究,H2O2直接合成用Pd负载型催化剂的研究取得了重大进展,主要集中在单一负载型Pd催化剂、Pd催化剂活性组分改性、载体改性的研究上。可以归结为以下几个方面:
(1)Pd催化剂活性组分Pd的颗粒尺寸、分散度、能量位点以及载体表面酸性对H2O2直接合成性能有明显影响。Pd颗粒尺寸小,分散度高,提供的活性位多,单位时间、单位催化剂上吸附与活化的H2和O2多,催化活性较好;但Pd颗粒尺寸较小,Pd位点能量过高,容易活化吸附在Pd表面的O2分子和H2O2分子中O-O键,使其容易断裂,吸附态分子(O2)a解离为吸附态原子Oa,降低合成H2O2选择性,H2O2分子解离为H2O,降低H2O2稳定性。
(2)贵金属Au或Pt、非贵金属对Pd催化剂活性组分改性,利于降低活性组分Pd颗粒尺寸、减弱Pd-O键能,抑制吸附态的分子氧(O2)a解离为吸附态的原子氧Oa,从而抑制合成H2O的反应,提高H2O2选择性。
(3)酸或非金属如卤素离子、N元素等对Pd催化剂载体改性,利于抑制O2和H2O2中O-O键解离,提高H2O2稳定性或降低活性组分Pd颗粒尺寸,提高Pd分散度。
目前尚需解决的技术和理论难题,如关于如何通过实验和理论得到Pd位点能量数据的相关研究还太少,尤其是关于经贵金属和非贵金属改性后的Pd催化剂上Pd位点或合金位点能量的计算鲜有报道,以致不能有效地解决当Pd颗粒尺寸更小时,分散度更大,活性位更多,而催化活性反而更低的原因,这有待于进一步深入研究。其次,欠缺载体自身性能(晶型、结构等)与催化活性的相关研究,考察载体自身结构及表面酸性对直接法合成H2O2的动力学研究仍具有重大意义。
[1]Henke H,Weber W.Manufacture of hydrogen peroxide [P].US:1108752A,1914.
[2]Gosser L W,Schwartz J T.Hydrogen peroxide production methodusingplatinum/palladiumcatalysts[P].US: 4832938A,1989.
[3]Liu Q S,Chris Bauer J,Schaak E,et al.Supported palladium nanoparticles:An efficient catalyst for the direct formation of H2O2from H2and O2[J].Angew Chem,2008,120:6317-6320.
[4]Dittmeyer R,Grunwaldt J D,Pashkova A.A review of catalyst performance andnovelreactionengineering concepts in direct synthesis of hydrogen peroxide[J]. Catal Today,2015,248:149-159.
[5]Edwards J K,Freakley S J,Lewis R J,et al.Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen[J].Catal Today,2015,248:3-9.
[6]Li J,Ishihara T,Yoshizawa K.Theoretical revisit of the direct synthesis of H2O2on Pd and Au@Pd surfaces:A comprehensive mechanistic study[J].J Phys Chem C, 2011,115:25359-25367.
[7]Todorovic R,Meyer R J.A comparative density functional theory study of the direct synthesis of H2O2on Pd,Pt and Au surfaces[J].Catal Today,2011,160:242-248.
[8]Ham H C,Hwang G S,Han J,et al.On the role of Pd ensembles in selective H2O2formation on Pd@Au Alloys [J].J Phys Chem C,2009,113:12943-12945.
[9]Staykov A,Kamachi T,Shihara T,et al.Theoretical study of the direct synthesis of H2O2on Pd and Pd/Au surfaces [J].J Phys Chem C,2008,112:19501-19505.
[10]Li J,Staykov A,Ishihara T,et al.Theoretical study of the decomposition and hydrogenation of H2O2on Pd and Au@Pd surfaces:Understanding toward high selectivity of H2O2synthesis[J].J Phys Chem C,2011,115:7392-7398.
[11]Li J,Yoshizawa K.Mechanistic aspects in the direct synthesis of hydrogen peroxide on PdAu catalyst from first principles[J].Catal Today,2015,248:142-148.
[12]DeguchiT,YamanoH,IwamotoM.Kineticand mechanistic studies on direct H2O2synthesis from H2and O2catalyzed by Pd in the presence of H+and Br-in water:A comprehensive paper[J].Catal Today,2015, 248:80-90.
[13]Deguchi T,Iwamoto M.Reaction mechanism of direct H2O2synthesis from H2and O2over Pd/C catalyst in water with H+and Br ions[J].J Catal,2011,280:239-246.
[14]陈诵英,孙予罕,丁云杰,等.吸附与催化[M].郑州:河南科学技术出版社,2001:136.
[15]Ye Y J,Chun J Y,Park S Y,et al.A study of the palladium size effect on the direct synthesis of hydrogen peroxide from hygrogen and oxygen using highly uniform palladium nanoparticles supported on carbon[J].Korean J Chem Eng,2012,29:1115-1118.
[16]Menegazzo F,Signoretto M,Frison G,et al.When high metal dispersion has a detrimental effect:Hydrogen peroxidedirectsynthesisunderverymildand nonexplosive conditions catalyzed by Pd supported on silica[J].J Catal,2012,290:143-150.
[17]GhediniaE,MenegazzoF,SignorettoaM,etal. Mesoporous silica as supports for Pd-catalyzed H2O2direct synthesis:Effect of the textural properties of the support on the activity and selectivity[J].J Catal,2010, 273:266-273.
[18]Tian P F,Ouyang L K,Xu X C,et al.Density functional theory study of direct synthesis of H2O2from H2and O2on Pd(111),Pd(100),and Pd(110)surfaces[J].Chin J Catal, 2013,34:1002-1012.
[19]Kim S,Lee D W,Lee K Y.Direct synthesis of hydrogen peroxide from hydrogen and oxygen over single-crystal cubic palladium on silica catalysts[J].J Mol Catal A, 2014,383-384:64-69.
[20]Kim S,Lee D W,Lee K.Shape-dependent catalytic activity of palladium nanoparticles for the direct synthesis of hydrogen peroxide from hydrogen and oxygen[J].J Mol Catal A,2014,391:48-54.
[21]Hua B Z,Deng W P,Rong S,et al.Carbon-supported palladium catalysts for the direct synthesis of hydrogen peroxide from hydrogen and oxygen[J].J Catal,2014, 319:15-26.
[22]PashkovaA,DittmeyerR,KaltenbornN,etal. Experimentalstudyofporoustubularcatalytic membranes for direct synthesis of hydrogen peroxide[J].J Chem Eng,2010,165:924-933.
[23]Lee H J,Kim S,Lee D W,et al.Direct synthesis of hydrogen peroxide from hydrogen and oxygen over a Pd core-silica shell catalyst[J].Catal Commun,2011,12: 968-971.
[24]Park S Y,Lee J,Song J H,et al.Direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd/ HZSM-5 catalysts:Effect of Brösted acidity[J].J Mol Catal A,2012,363-364:230-236.
[25]Park S Y,Seo J G,Jung J C,et al.Direct synthesis of hydrogenperoxidefromhydrogenandoxygenover palladium catalysts supported on TiO2-ZrO2mixed metal oxides[J].Catal Commun,2009,10:1762-1765.
[26]Wang L,Bao S G,Yi J H,et al.Preparation and properties of Pd/Ag composite membrane for direct synthesis of hydrogen peroxide from hydrogen and oxygen [J].Appl Catal B,2008,79:157-162.
[27]Abate S,Melada S,Centi G,et al.Performances of Pd-Me (Me=Ag,Pt)catalysts in the direct synthesis of H-O-on catalytic membranes[J].Catal Today,2006,117:193-198.
[28]Choudhary V R,Samanta C,Choudhary T V.Direct oxidation of H2to H2O2over Pd-based catalysis:influence of oxidation state,support and metal additives[J].Appl Catal A,2006,308:128-133.
[29]Bernardotto G,Menegazzo F,Pinna F,et al.New Pd-Pt and Pd-Au catalysts for an efficient synthesis of H2O2from H2and O2under very mild condition[J].Appl Catal A,2009,358:129-135.
[30]Edwards J K,Hutchings G J.Palladium and goldpalladium catalysts for the direct synthesis of hydrogen peroxide[J].Angew Chem,2008,47:9192-9198.
[31]Menegazzo F,Signoretto M,Manzoli M,et al.Influence of thepreparationmethodonthemorphologicaland composition properties of Pd-Au/ZrO2catalysts and their effect on the direct synthesis of hydrogen peroxide from hydrogen and oxygen[J].J Catal,2009,268:122-130.
[32]Gudarzi D,Simakova O A,Hernandea Carucci J R,et al. Direct synthesis of H2O2from H2and O2over carbon supported Au,Pd and Au-Pd/C bimetallic catalysts[J]. Chem Eng Trans,2010,21:925-930.
[33]Edwin N.Ntainjua M P,Simon J F,et al.Direct synthesis ofhydrogenperoxideusingAu-Pd-exchangedand supportedheteropolyacidcatalystsatambient temperature using wateras solvent[J].Green Chem,2012, 14:170-181.
[34]Xu J,Ouyang L K,Da G J,et al.Pt promotional effects on Pd-Pt alloy catalysts for hydrogen peroxide synthesis directly from hydrogen and oxygen[J].J Catal,2012,285: 74-82.
[35]García T,Agouram S,Ana D,et al.Enhanced H2O2production over Au-rich bimetallic Au-Pd nanoparticles on ordered mesoporous carbons[J].Catal Today,2015, 248:48-57.
[36]Ouyang L K,Da G J,Tian P F,et al.Insight into active sitesofPd-Au/TiO2catalystsinhydrogenperoxide synthesis directly from H2and O2[J].J Catal,2014,311: 129-136.
[37]Davood G,Warin R,Ilkka T,et al.Promotional effects of Au in Pd-Au bimetallic catalysts supported on activated carbon cloth(ACC)for direct synthesis of H2O2from H2and O2[J].Catal Today,2015,248:58-68.
[38]Edwards J K,Solsona B,Landon P,et al.Direct synthesis of hydrogen peroxide from H2and O2using Au-Pd/Fe2O3catalysts[J].J Mater Chem,2005,15:4595-4600.
[39]Edwards J K,Thomas A,Solaona B E,et al.Comparison of supports for the direct synthesis of hydrogen peroxide from H2and O2using Au-Pd catalysts[J].Catal Today, 2007,122:397-402.
[40]Pritchard J,Kesavant L,Piccininii M,et al.Direct synthesisofhydrogenperoxideandbenzylalcohol oxidationusingAu-Pdcatalystspreparedbysol immobilization[J].Langmuir,2010,26:16568-16577.
[41]安红强,王桂赟,王延吉,等.过渡金属改性的Pd/TS-催化氢、氧直接合成H2O2的性能[J].无机化学学报,2010,26 (3):405-412.
[42]杜旭,潘红艳,阁世媚,等.氢氧直接合成过氧化氢用PdO/γ-Al2O3催化剂过渡金属改性研究[J].无机盐工业, 2013,45(10):49-52.
[43]笪国进,欧阳李科,徐晶,等.吡啶改性Pd/SiO2催化剂用于H2和O2直接合成H2O2[J].化工学报,2013,64(2): 561-567.
[44]Choudhary V R,Samanta C,Jana P.Hydrogenation of hydrogen peroxide over palladium/carbon in aqueous acidic medium containing different halide anions under static/flowing hydrogen[J].Ind Eng Chem Res,2007,46: 3237-3242.
[45]Samanta C,Choudhary V R.Direct formation of H2O2from H2and O2and decomposition/hydrogenation of H2O2in aqueous acidic reaction medium over halide-containing Pd/SiO2catalytic system[J].Catal Commun,2007,8: 2222-2228.
[46]Samanta C,Choudhary V R.Direct oxidation of H2to H2O2over Pd/Ga2O3catalyst under ambient conditions: Influence of halide ions added to the catalyst or reaction medium[J].Appl Catal A,2007,326:28-36.
[47]Choudhary V R,Jana P.Direct H2-to-H2O2oxidation over highly active/selective Br-F-Pd/Al2O3catalyst in aqueous acidic medium:Influence of process conditions on the H2O2formation[J].Appl Catal A,2009,352:35-42.
[48]Choudhary V R,Jana P.Direct oxidation of H2to H2O2over Br and F-promoted Pd/Al2O3in aqueous acidic medium:Influence of the concentration of Br and F and the method of incorporation of the two halogens in the catalyst on their beneficial synergetic effect on the netH2O2formation[J].Appl Catal A,2007,329:79-85.
[49]Choudhary V R,Samanta C,Jana P.Decomposition and/ or hydrogenation of hydrogen peroxide over Pd/Al2O3catalyst in aqueous medium:Factors affecting the rate of H2O2destruction in presence of hydrogen[J].Appl Catal A,2007,332:70-78.
[50]Choudhary V R,Samanta C,Jana P.Formation from direct oxidation of H2and destruction by decomposition/ hydrogenation of H2O2over Pd/C catalyst in aqueous medium containing different acids and halide anions[J]. Appl Catal A,2007,317:234-243.
[51]Choudhary V R,Samanta C,Choudhary T V.Factors influencing decomposition of H2O2over supported Pd catalyst in aqueous medium[J].J Mol Catal A,2006,260: 115-120.
[52]Choudhary V R,Ingole Y V,Samanta C,et al.Direct oxidation of hydrogen to hydrogen peroxide over Pd (or PdO)/Al2O3in aqueous reaction medium:Influence of different acids and halide anions in reaction medium on formation and destruction of H2O2[J].Ind Eng Chem Res, 2007,46:8566-8573.
[53]Edwards J K,Carley A F,Herzing A A,et al.Direct synthesis of H2O2from H2and O2over gold,palladium, and gold-palladium catalysts supported on acid-pretreated TiO2[J].Angew Chem,2009,48:8512-8515.
[54]Edwards J K,Parker S F,Pritchard J,et al.Effect of acid pre-treatment on AuPd/SiO2catalysts for the direct synthesis of hydrogen peroxide[J].Catal Sci Technol, 2013,3:812-818.
[55]Edwards J K,Solsonal B,Edwin N N,et al.Switching off hydrogen peroxide hydrogenation in the direct synthesis process[J].Science,2009,323:1037-1041.
[56]Park S Y,Baeck S H,Kim T J,et al.Direct synthesis of hydrogenperoxidefromhydrogenandoxygenover palladium catalyst supported on SO3H-functionalized mesoporous silica[J].J Mol Catal A,2010,319:98-107.
[57]Park S Y,Park D R,Choi J H,et al.Direct synthesis of hydrogenperoxidefromhydrogenandoxygenover palladium catalyst supported on H3PW12O40-incorporated MCF silica[J].J Mol Catal A,2011,336:78-86.
[58]Park S Y,Choi J H,Kim T J,et al.Direct synthesis of hydrogenperoxidefromhydrogenandoxygenover Pd/CsXH3-XPW12O40/MCF(X=1.7,2.0,2.2,2.5,and 2.7) catalysts[J].J Mol Catal A,2012,353-354:37-43.
[59]Park S Y,Choi J H,Kim T J,et al.Direct synthesis of H2O2from H2and O2over Pd catalyst supported on Cs2.5H0.5PW12O40-MCF silica[J].Catal Today,2012,185(1): 162-167.
[60]Samanta C,Choudhary V R.Direct synthesis of H2O2from H2and O2and decomposition/hydrogenation of H2O2in an aqueous acidic medium over halide-modified Pd/Al2O3catalysts[J].Appl Catal A,2007,330:23-32.
[61]Abate S,Arriog R,Schuster M E,et al.Pd Nanoparticles supported on N-doped nanocarbon for the direct synthesis of H2O2from H2and O2[J].Catal Today,2010,157:280-285.
[62]Park S Y,Kim T J,Chung Y M,et al.Direct synthesis of hydrogenperoxidefromhydrogenandoxygenover insoluble Pd0.15M2.5H0.2PW12O40(M=K,Rb,and Cs)heteropoly acid catalysts[J].Res Chem Int,2010,36:639-646.
An overview of recent developments on palladium based catalysts for direct synthesis of hydrogen peroxide from hydrogen and oxygen under atmospheric pressure
TIAN Min1,PAN Hong-yan1,LIN Qian2,CHEN Zhi-chao1,ZHAO Min1,CHEN Zheng1
(1.School of Chemical Engineering,Guizhou University,Guiyang 550025,China; 2.Key Laboratory of Guizhou Province for Green Chemical Industry and Clean Energy Technology,Guiyang 550025,China)
Recent developments on the palladium based catalysts for direct synthesis of hydrogen peroxide from hydrogen and oxygen under atmospheric pressure were summarized,including the single palladium supported catalysts,the modification of active components on palladium catalysts by doping with other metals and/or non-metals,the modification of supports by pretreatment with acids and/or other species.The effects of palladium catalyst properties,including particle size,dispersion and energy sites of palladium and the surface acidity of supports,on the catalytic activity for direct synthesis of hydrogen peroxide were discussed.It was indicated that the development direction of palladium catalysts is still to improve the yield and concentration of hydrogen peroxide to meet the needs of industrial applications by strengthening the main reaction and restraining the side reactions.
hydrogen peroxide;direct synthesis;palladium catalyst;modification
TQ426.94;TQ123.6
:A
:1001-9219(2016)01-91-08
2015-04-30;基金项目:国家自然科学基金项目(21366008),黔科合JZ字[2014]2008号,黔科合J字[2012] 2152号;作者简介:田敏(1991-),女,硕士研究生,电邮mtian1015@163.com;*
潘红艳(1983-),女,副教授,电邮cepanhongyan@163.com;林倩(1962-),女,教授,电邮linqian@126.com。
