园艺作物基因组测序研究进展
2016-04-11倪晓鹏高志红
倪晓鹏++高志红
摘要:回顾了园艺作物全基因组测序的发展历程,介绍3代测序技术的特点和应用现状。总结了葡萄、番木瓜、草莓等果树,黄瓜、白菜、番茄等蔬菜以及莲、康乃馨等花卉在内的25种园艺作物基因组测序简况。重点介绍了全基因组测序在基因注释、比较基因组学研究、重测序、全基因组关联分析、转录组学、SNP芯片开发等方面的应用。最后讨论了全基因测序研究的难点和今后的研究方向。
关键词:园艺作物;基因组测序;基因组学
中图分类号: Q78文献标志码: A 文章编号:1002-1302(2016)02-0009-04
园艺业是农业种植业的重要组成部分,对丰富人类营养和美化、改善人类生存环境有重要意义。我国是世界园艺大国,园艺是我国农业的重要组成部分,其中蔬菜播种面积由1990年的600万hm2增加到2013年的2 100万hm2,产量由1990年的1.95亿t增加到2013年的7.35亿t[1]。种植面积和产量分别占世界的43%和49%,居世界第一位。随着动植物全基因组测序的不断发展,2001年由美国能源部推动的人类基因组工作草图的发表被认为是人类基因组计划成功的里程碑[2]。破译人类遗传信息,将对医学、生物学乃至整个生物科学产生不可估量的影响。这一计划促成了大批动植物全基因测序的完成,也有望从基因组水平上分析基因的结构、组成、调控和物种进化,从而避免传统分子生物学带来的种种弊端,大大促进全基因组测序技术在园艺作物上的应用发展。
1全基因测序的发展历程及其在园艺植物测序中的应用
距世界第一例模式植物拟南芥全基因组测序发表已有十余年[3],在此期间测序技术飞速发展,每年发表的测序文章都稳定增加。拟南芥的测序就是基于第一代测序技术Sanger测序完成的,运用了链终止和断裂技术。第一代测序技术已经规模化,且具有测序读长较长、测序准确率高等特点,但是由于其时间长、成本高、通量低等缺点而无法满足现在实验需求。
2004年,美国国家人类基因组研究所(NHGRI)发起了一项融资计划[4],目标是在未来10年将人类基因组测序的费用减少到1 000美元,这刺激了商业化的新一代测序(next generation sequencing,NGS)技术的快读萌芽。第二代测序技术大幅提升基因组测序的输出与成本比,相较第一代测序技术涵盖范围更加广泛,可以同时测定多个平行DNA片段,从而输出更多测序阅读量,但一般长度较短、质量低。第一个NGS技术是罗氏公司(Roche)公司的454焦磷酸测序法[5],Solexa和Illumina测序平台在1年后也进行商业化应用。第二代测序技术具有成本低、单次数据量大、消耗时间短等特点,故又被称为第二代高通量测序技术,并渐渐成为大规模全基因组测序技术的主导。
Roche 454测序平台是基于焦磷酸测序,由于低数据率和相对较短的读取,最初适用于细菌基因组测序,随后技术改进,利用Roche 454与Sanger测序结合起来运用于更复杂的基因组测序,完成苹果基因组测序[6],取代Sanger测序作为主要数据源完成可可基因组[7]和甜瓜[8]基因组测序。Illumina 公司提供的Solexa系统比Roche 454系统数据产出量多且花费较少的费用。自推出以来,读取长度获得明显改善,并成功运用到黄瓜的测序工作中[9]。
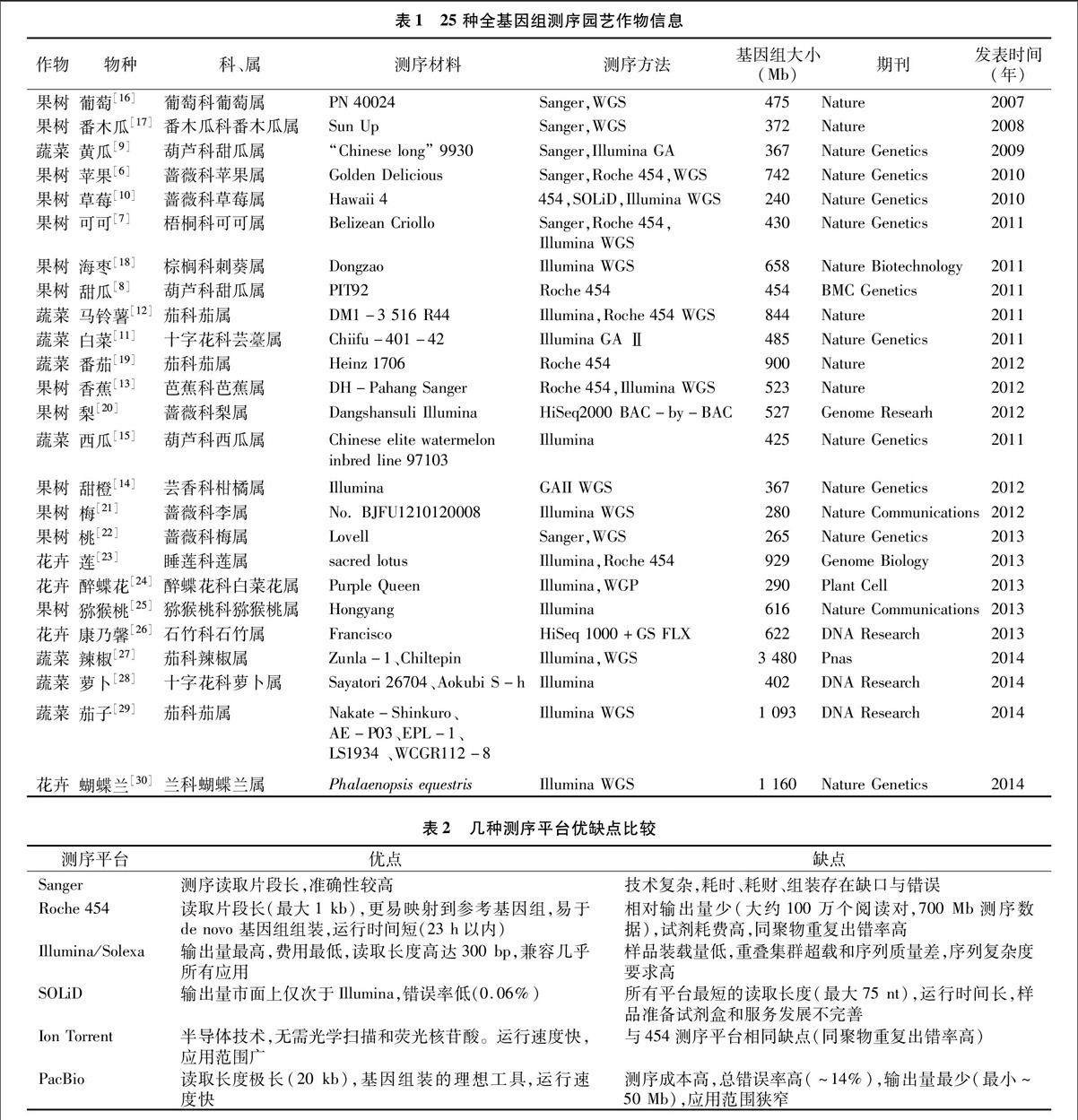
野生草莓[10]作为第一个采用Roche 454、Illumina、SOLiDTM三大平台共同完成测序的植物,标志着测序技术向多平台合作而不是独立运行的模式发展。近几年来,Illumina测序成为第二代测序平台的主导,截至2014年11月,已为多种园艺作物如中国白菜[11]、马铃薯[12]、香蕉[13]、橙[14]和西瓜[15]基因组测序(表1)。
近年来,新一代测序平台已经出现并被称为“第三代测序技术”,与第二代测序方法相比有着进一步模式转变,有2个突出特点,一是测序前不再需要PCR扩增,二是荧光或电流信号都可以在互补链加核苷酸的酶反应中被检测到。2种测序平台现在已经投入商业化运营,分别是Pacific Biosciences公司的PacBio RS平台(http://www. Pacificbio scie nces.com)和Ion Torrent 公司的Personal Genome平台 (Life Technologies,http://www.iontorren t.com)。PacBio测序平台使用实时零模式波导检测单个DNA聚合酶的活性[31],大规模并行单分子实时(SMRT)测序保证了高通量测序。其突出特点是序列读长,经报道其单程长读取对的准确率为84.2%~97.8%[32],相较于第二代测序技术,第三代测序技术可以读取更长序列、提高测序通量、节省试剂成本,但其错误率比第二代测序技术高,3代测序技术优缺点比较见表2。
2全基因组测序的应用
2.1基因注释
基因注释是利用生物信息学方法给测序完成的物种序列附上生物学信息的过程,通过识别不编码蛋白质的基因片段,识别基因上的元素(基因预测),给元素附上生物学信息的手段进行重复序列的识别、非编码RNA的预测、基因结构预测和基因功能注释,发现与农艺性状相关基因,例如花期调控、植物生长习性、耐寒性、休眠、果实性状与品质等。Fukuoka等通过鉴定苹果(Malus×domestica)的146个MADS-box基因,与拟南芥和水稻MADS-box基因聚成6个亚组,预测MADS-box基因在17条染色体上的密度,在根、茎、叶、花组织和果实发育的5个阶段进行了分析,表明MADS-box基因参与了苹果的生理和发育过程的各个方面[33]。Yu等将超过6 000 个茄子组织和处理组的cDNA克隆组成1个含有16 245 个独特序列的单基因组,对单基因组进行测序并采用SAGE策略对茄子单基因集在转录组中的功能分析,相当数量的短序列标签被成功注释[34]。
2.2比较基因组学研究
比较基因组学研究是基于基因组图谱和测序技术,对已知的基因和基因组结构进行比较以了解相关基因的功能、表达机制和物种亲缘关系。
番茄测序小组完成了对栽培番茄及其近缘野生种醋栗番茄全基因组的精细序列分析。在解码的番茄基因组中共鉴定出 34 727 个基因,其中97.4% 的基因已经精确定位到染色体上。通过比较基因组分析发现了番茄果实进化和发育的基因组学基础,番茄基因组经历的2次三倍化使基因家族产生了特异控制果实发育及营养品质的新成员[19]。Zhang等研究发现在梅花基因组中存在和抗病相关的PR基因家族,由PR编码的蛋白质可介导植物产生防御反应,以此来抵抗病害和不良环境。例如:梅花基因组中存在的[WTBX][STBX]PR10[WTBZ][STBZ]与梅花的叶和根抗盐度、抗干旱和抗病菌的机制相关[21]。
2.3重测序
通过基因组重测序,可以对栽培种和野生种基因组之间的差异进行比较,从而揭示物种起源以及驯化过程,为鉴定有价值的遗传资源以及园艺作物育种提供重要参考。
Qi等对115个黄瓜品系进行了深度重测序,鉴定出112个假定的驯化延伸,其中1个区域包含着参与黄瓜果实苦味消失过程的1个基因,以及1个重要的驯化特性,也调查了栽培品种中分离度的基因组基础,在β-胡萝卜素羟化酶发现了一个自然遗传变异,可以用来培育含有更高营养价值的黄瓜,揭露黄瓜进化的遗传历程,为未来遗传育种提供了保证[35]。
2.4全基因组关联分析
全基因组关联分析(genome-wide association study,GWAS)是应用基因组中数以百万计的单核苷酸多态性(single nucleotide ploymorphism,SNP)作为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。
Kenta等为了研究栽培番茄遗传和表型多样性的关系,重测序6个栽培番茄品系,基于全基因连锁不平衡分析表明,连锁不平衡的延长依赖于染色质的性质,全基因组关联分析表明所鉴定的SNP与农艺重要的性状有着密切的关系,3个基因(FAS、SP、U)被发现与性状变异相关,证明通过重测序得到的大量SNP进行全基因组关联分析可以鉴别特异性位点,表明栽培番茄在基因组学和遗传学发展上又迈进了一大步[36]。
2.5与转录组数据比较分析
转录组研究是基因功能及结构研究的基础和出发点,是全基因组测序完成后首先要面对的问题。最近科学家们将高通量测序技术应用于转录组分析开发出了RNA测序技术(RNA-SEq),该技术能够在全基因组范围内检测基因表达情况,进行差异基因筛选分析。由于RNA-SEq技术具有通量高、可重复性好、检测范围宽、定量准等特点,已经广泛应用于细菌、拟南芥、水稻和人类等生物转录组的研究。
Zenoni等利用RNA-Seq技术对葡萄栽培种Corvina浆果发育3个阶段的转录组变化进行了研究,对比Pinot Noir 40024参考基因组,分析测量基因表达水平,共检测到17 324个基因在果实发育过程中表达,其中6 695个在不同阶段会特异性表达,这表明了基因功能的多样性与表达特异性以及转录组的高度复杂性[37]。
2.6SNP芯片的开发
单核苷酸多态性(SNP)根据其在基因中的位置,可以分为基因编码区、基因非编码区、基因间隔区(基因之间的区域),在基因组中是多态性最丰富的DNA分子标记,具有数量众多、分布均匀、分型方便等特点,它可以识别遗传变异和关联的表型基因分型。
Chagné等利用生物信息学工具,从包含苹果品系中350 000 条序列的EST数据库中开发SNP标记,结果识别了71 482个假定SNP分子标记。设计了464个PCR引物对,对PCR产物进行测序,重新获得的SNP标记映射到苹果参考基因(Royal Gala×A689-24杂交系和Malling 9×Robusta 5)SNP基因分型采用高分辨率熔解 (HRM)技术,共包含210个编码SNP的93个新标记被成功映射到参考图谱。此方法为使用数量性状定位(QTL)技术了解重要农艺性状和连锁不平衡分析的基因调控提供了借鉴,也成为物理和遗传图谱结合的有效标记[38]。
3全基因组测序存在的挑战
测序任何基因组的挑战包括倍性和杂合度。与动物的基因组相比,植物基因组具有巨大的基因组大小、高度重复序列和全基因组或者片段基因组复制。
第二代测序平台Roche 454、 Illumina和SoLiD不仅吞吐量显著增加,还减少了错误率,增加了阅读长度,使大范围物种测序都能采用测序平台经济地进行。然而,植物基因组的独特性给测序带来了许多挑战,如他们的重复特性,装配完整基因组是具有挑战性的,这是由于植物基因组转座因子的高拷贝数和扩张特性决定的。例如,玉米基因组测序通过BAC-by-BAC方法,基因组中85%含有转座元件测序[39]。植物基因组中全基因组、节段和串联重复的频率[40]也给旁系同源种类带来组装问题,如果是最近一次复制,其序列同源性会高。因此,尽管第二代测序平台能够测序一大部分基因组,但迄今为止的全基因组测序方案还是缺乏足够的代表性,质量评估在组装过程中还会丢失部分基因。
另一个问题是,不是所有植物品种都是纯合二倍体自交系,尽管使用WGS方法为杂合组织测序是可行的。然而高杂合度,包括交叉和无性繁殖的物种,如马铃薯,会阻碍WGS组装。为了解决葡萄基因组(高度杂合二倍体)问题,Jaillonet等[JP3]采用了高度近交的品种,减少了杂合程度,进而才完成了测序。使用长阅读可以提高一个基因组中组装分离单体型的能力[16]。
植物倍性也是在植物denovo测序和组装的一个巨大挑战,其结果取决于物种是一个同源多倍体是异源多倍体,迄今为止,所有多倍体测序依赖于倍性降低或者染色体的物理分离。例如,绝大多数马铃薯品系是四倍体,最初是取杂合二倍体马铃薯基因 (RH89-039-16)进行测序从而避免同源染色体之间的高度杂合性,最后利用1个特殊的马铃薯基因型,1个加倍的单倍体 (DM1-3 516 R44),含有纯合的12条染色体作为参考马铃薯基因组[12]。栽培草莓(Fragaria ananassa)是来自4个不同祖先的异源八倍体,野生草莓(Fragaria vesca)是二倍体,被用来测序是为了避免测序多个基因组带来的困难[10]。
4全基因组测序的展望
为了面对世界基因组测序技术发展如此迅猛的挑战,大量全基因组测序数据亟待深度挖掘,应制定长远的战略性基因组学研究计划,不仅局限于栽培物种,更应该深度开发我国重要野生近缘物种的测序,促进重要基因资源的挖掘、保护和利用。摆脱先前依赖外观表现型而转入到基因型依赖型的研究当中去,从单一基因研究深入到全基因组关联分析研究中。大力推动我国基因组学在基因克隆与分子育种领域的应用研究,提高园艺作物育种能力和水平。加强与转录组学、代谢组学、蛋白质组学和降解组学的相关研究,促进基因组学生物信息的共享与利用。加强生物信息学教育的投入,并将其应用于实践中。随着测序技术的不断发展,相信在不久的将来园艺作物全基因组测序将进入快速发展的阶段,为世界园艺产业带来巨大贡献。
[HS2][HT8.5H]参考文献:[HT8.SS]
[1][ZK(#]中华人民共和国农业部.中国农业统计资料:2013[M]. 北京:中华人民共和国农业部,2014.
[2]Collins F S,Morgan M,Patrinos A. The human genome project:lessons from large-scale biology[J]. Science,2003,300(5617):286-290.
[3]Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J]. Nature,2000,408(6814):796-815.
[4]Schloss J A. How to get genomes at one ten-thousandth the cost[J]. Nature Biotechnology,2008,26(10):1113-1115.
[5]Margulies M,Egholm M,Altman W E,et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature,2005,437(757):376-380.
[6]Velasco R,Zharkikh A,Affourtit J,et al. The genome of the domesticated apple (Malus × domestica Borkh.)[J]. Nature Genetics,2010,42(10):833-839.
[7]Argout X,Salse J,Aury J M,et al. The genome of Theobroma cacao[J]. Nature Genetics,2011,43(2):101-108.
[8]Rodríguez-Moreno L,González V M,Benjak A,et al. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin[J]. BMC Genomics,2011,12:424.
[9]Huang S,Li R,Zhang Z,et al. The genome of the cucumber,Cucumis sativus L.[J]. Nature Genetics,2009,41(12):1275-1281.
[10][ZK(#]Shulaev V,Sargent D J,Crowhurst R N,et al. The genome of woodland strawberry (Fragaria vesca)[J]. Nature Genetics,2011,43(2):109-116.
[11]Wang X,Wang H,Wang J,et al. The genome of the mesopolyploid crop species Brassica rapa[J]. Nature Genetics,2011,43(10):1035-1039.
[12]Potato Genome Sequencing Consortium,Xu X,Pan S,et al. Genome sequence and analysis of the tuber crop potato[J]. Nature,2011,475(7355):189-195.
[13]Dhont A,Denoeud F,Aury J M,et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants[J]. Nature,2012,488(7410):213-217.
[14]Xu Q,Chen L L,Ruan X,et al. The draft genome of sweet orange (Citrus sinensis)[J]. Nature Genetics,2013,45(1):59-66.
[15]Guo S,Zhang J,Sun H,et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions[J]. Nature Genetics,2013,45(1):51-58.
[16]Jaillon O,Aury J M,Noel B,et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J]. Nature,2007,449(7161):463-467.
[17]Ming R,Hou S,Feng Y,et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus)[J]. Nature,2008,452(7190):991-996.
[18]Al-Dous E K,George B,Al-Mahmoud M E,et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera)[J]. Nature Biotechnology,2011,29(6):521-527.
[19]Sato S,Tabata S,Hirakawa H,et al. The tomato genome sequence provides insights into fleshy fruit evolution[J]. Nature,2012,485(740):635-641.
[20][JP3]Wu J,Wang Z,Shi Z,et al. The genome of the pear (Pyrus bretschneideri Rehd.)[J]. Genome Research,2013,23(2):396-408.
[21]Zhang Q,Chen W,Sun L,et al. The genome of Prunus mume[J]. Nature Communications,2012,3:1318.
[22]International Peach Genome Initiative,Verde I,Abbott A G,et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity,domestication and genome evolution[J]. Nature Genetics,2013,45(5):487-494.
[23]Ming R,Vanburen R,Liu Y,et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.)[J]. Genome Biology,2013,14(5):R41.
[24]Cheng S,van den Bergh E,Zeng P,et al. The tarenaya hassleriana genome provides insight into reproductive trait and genome evolution of crucifers[J]. The Plant Cell,2013,25(8):2813-2830.
[25]Huang S,Ding J,Deng D,et al. Draft genome of the kiwifruit Actinidia chinensis[J]. Nature Communications,2013,4:2640.
[26]Yagi M,Kosugi S,Hirakawa H,et al. Sequence analysis of the genome of carnation (Dianthus caryophyllus L.)[J]. DNA Research:an International Journal for Rapid Publication of Reports on Genes and Genomes,2014,21(3):231-241.
[27]Qin C,Yu C,Shen Y,et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization[J]. Proceedings of the National Academy of Sciences of the United States of America,2014,111(14):5135-5140.
[28]Kitashiba H,Li F,Hirakawa H,et al. Draft sequences of the radish (Raphanus sativus L.) genome[J]. DNA Research:an International Journal for Rapid Publication of Reports on Genes and Genomes,2014,21(5):481-490.
